
一种新的对1-H-1,2,4-三氮唑的高效液相色谱定量分析方法
2018-03-04 12:51:46
四川化工 2018年6期
(四川省化学工业研究设计院,四川成都,610041)
1-H-1,2,4-三氮唑是一种含氮杂环化合物,作为染料、医药、农药工业的重要中间体,在多种化工合成工艺中具有重要作用。三氮唑的常用分析方法有滴定法、气谱分析法和液谱分析法。液谱法相较于前两种方法具有易于操作和重现性好等特点。但由于三氮唑极性较强,极易溶于水,在传统反相色谱中,保留时间太短,当样品中有其他强极性杂质时,就给定量分析带来很大难度。
亲水作用色谱(HILIC),是上世纪九十年代开始发展的一种色谱技术,用来改善在反相色谱中强极性和亲水性物质保留时间较短、分离能力较差的问题。该技术利用强极性的固定相和高比例有机相的流动相,来达到此目的。因此,针对三氮唑分析工作中遇到的问题,我们尝试采用HILIC色谱柱来解决。
1 实验部分
1.1 仪器
高效液相色谱仪:Agilent 1100,紫外检测器;色谱柱:博纳艾杰尔 Venusil HILIC, 4.6×250mm,5μm不锈钢柱;紫外分光光度计:岛津UV-1800;天平:Mettler Toledo AL204-IC。
1.2 试剂
水:二次蒸馏水;乙腈:色谱纯;三氮唑标品:99%;试样:三氮唑(本单位)。
1.3 分析条件
流动相:乙腈:水 = 90:10 (V/V);流速:1.0 mL/min;柱温:室温;检测波长:196nm;进样体积:20μL;保留时间:5.0min左右。
1.4 测定步骤
1.4.1 吸收波长的选择
用UV-1800紫外分光光度计扫描三氮唑标准品溶液,扫描范围在190-290nm。
1.4.2 标准溶液的配制
准确称取三氮唑标样一定量(精确至0.0001g)于50mL容量瓶中,用流动相定容,混合均匀后作为标准溶液备用。
1.4.3 试样溶液的配制
准确称取三氮唑样品一定量(精确至0.0001g)于50mL容量瓶中,用流动相定容,混合均匀后作为试样溶液备用。
1.4.4 测定
在前述条件下,待基线稳定后,连续注入标样溶液,待相邻两针峰面积变化<1.0%,开始按标样、试样、试样、标样的顺序进样。
以下为三氮唑标样及样品的液谱图(图1、2)。

图1 三氮唑标样

图2 三氮唑样品
1.4.5 计算
将测得的两针试样以及前后两针标样中三氮唑峰面积分别平均,试样中三氮唑的质量分数X(%)按下式计算:
X=A2·m1·PA1·m2×100%
式中:A1:标样三氮唑峰面积平均值;A2:样品三氮唑峰面积平均值;m1:所称取的标样三氮唑质量;m2:所称取的样品三氮唑质量;P:标样中的三氮唑质量分数。
2 结果与讨论
2.1 吸收波长的确定
通过紫外扫描,发现在波长为196nm时,三氮唑有最强吸收,且可能含有的杂质甲酰胺在此波长下也有较强吸收,因此选定196nm为检测波长。
2.2 流动相的确定
为确保三氮唑与杂质显著分离,选择不同比例的乙腈+水作为流动相进行分离。实验发现,当流动相为乙腈:水=90:10(V/V)时,分离效果理想,基线平稳,因此选择该比例为最佳流动相。
2.3 分析方法的线性相关性
称取不同质量的三氮唑标准品于6个50mL容量瓶中,用流动相定容,混匀。在前定条件下分别进行测定,以三氮唑进样质量为横坐标,以三氮唑峰面积为纵坐标,绘制线性关系曲线如图3,线性方程:y=343426x+1112.2,决定系数为r2=0.9991,相关系数为r=0.9995。
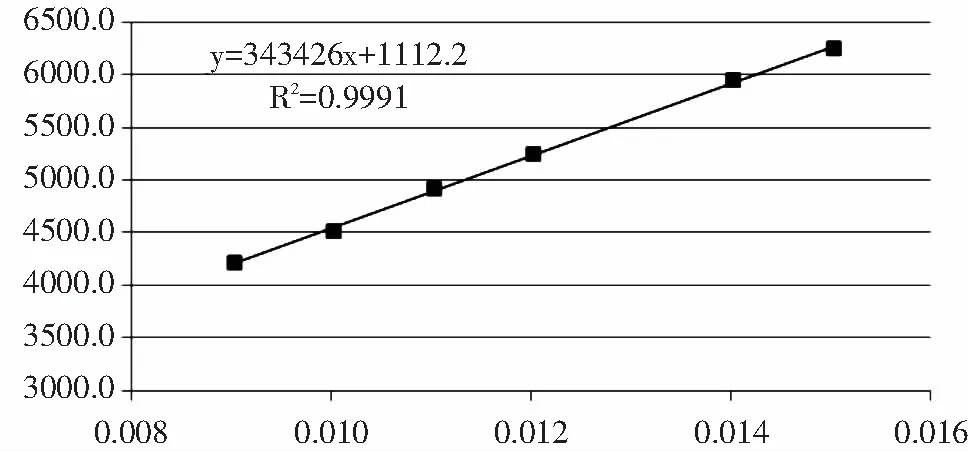
图3 三氮唑线性关系曲线图
2.4 方法精密度
分5次取同一批样品称量,采用前定条件进行分析,实验结果如表1。该方法测定三氮唑的标准偏差为0.22,变异系数为0.25%。证明该方法精密度良好。
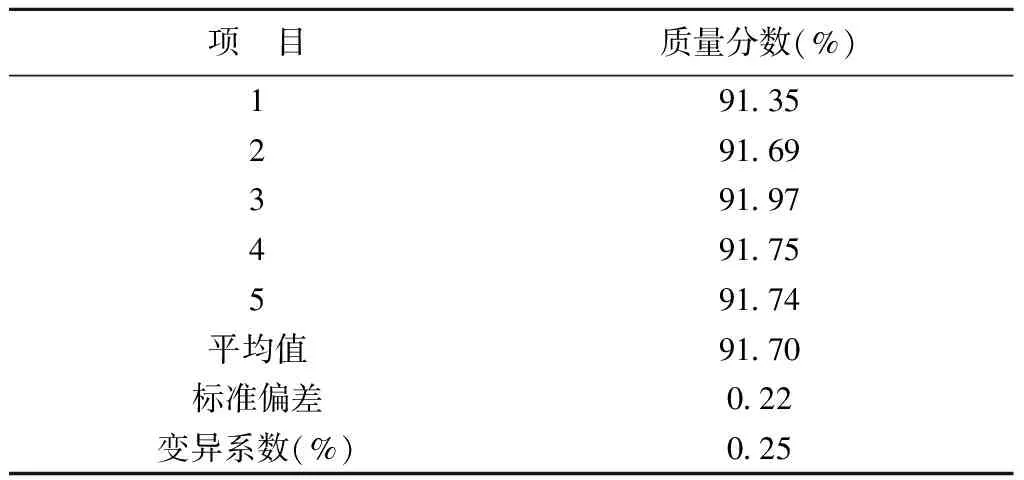
表1 三氮唑精密度实验结果
2.5 方法准确度
在已测得质量分数的三氮唑试样中,加入不同量的三氮唑标准品,配成5个已知样,在前定液谱条件下,定量分析、计算结果如表2,测得三氮唑平均回收率为97.57%,说明该方法准确度良好。
3 结论
如上所述,该方法测定三氮唑质量分数,简便、快速、准确,重现性、分离效果满足检验要求,相比原有反相液谱方法,更适用于企业质量检验。

表2 三氮唑准确度实验结果
猜你喜欢
特产研究(2022年6期)2023-01-17 05:06:16
理化检验-化学分册(2022年11期)2022-11-27 05:21:26
煤化工(2022年3期)2022-07-08 07:24:42
化工管理(2021年4期)2021-02-27 07:34:08
实用口腔医学杂志(2017年6期)2017-09-19 02:51:28
中国照明(2016年4期)2016-05-17 06:16:15
中国资源综合利用(2016年10期)2016-01-22 08:36:09
物理实验(2015年9期)2015-02-28 17:36:46
化工科技(2014年5期)2014-06-09 06:11:24
城市建设理论研究(2012年4期)2012-03-23 02:13:34
