
黑索金太赫兹吸收光谱仿真与形成机理的研究
2018-01-29 02:18祁乐融刘晓东
中北大学学报(自然科学版) 2017年6期
蔚 旋 , 王 高 , 祁乐融 , 韩 刚, 刘晓东
(1. 中北大学 信息与通信工程学院, 山西 太原 030051; 2. 中北大学 环境与安全工程学院, 山西 太原 030051; 3. 中北大学 仪器与电子学院, 山西太原 030051)
炸药分子对太赫兹波的吸收主要源于分子的低频振动模式, 其振动模式的位置和强度反映了分子的构型特征, 炸药的性能又与分子构型密切相关, 这使得分析炸药分子低频振动模式, 深入理解分子太赫兹吸收光谱特征及产生机理成为一项重要的研究课题, 可为该炸药相关性质的研究奠定基础[1]。 量子化学的发展以及一些仿真软件的出现, 为炸药太赫兹光谱解析提供了一种新的方法. 模拟计算炸药分子在太赫兹波段的振动模式, 可解析其太赫兹光谱的产生机理, 进而可分析该炸药的相关性质[2].
黑索金(RDX)是一种军用高能炸药, 与TNT相比, 其所需起爆能小, 爆炸威力更大, 被广泛应用于采矿与军事领域, 因此具有很大的研究价值. 在RDX太赫兹吸收光谱仿真及解析研究中, 文献[3] 计算得到了与实验结果吻合度很高的RDX太赫兹光谱, 但文献[4]与[5]只得到了3个与实验结果吻合的吸收峰, 存在峰位缺失和偏移, 没有得到准确的仿真光谱, 只是从单分子层面讨论了太赫兹吸收峰产生机理. 单体模型只能模拟分子内的振动情况, 但分子间的相互作用在物质太赫兹光谱中也起着重要的作用[6].
本文在借助量子化学软件Gaussian 09对RDX单分子太赫兹吸收光谱仿真的基础上, 进一步探究分子间作用对吸收光谱的影响, 对由氢键连接的RDX二聚体进行仿真, 比较了不同构型对RDX炸药太赫兹光谱仿真的影响. 同时, 从分子内的振动转动、 分子间作用解析了RDX太赫兹光谱产生机理.
1 基础理论
1.1 物质太赫兹波产生原理
一般地, 物质总是处于不停的运动中, 构成物质的分子和原子也保持一定的运动状态, 且每个状态都处在特定的能级[7]. 当物质被太赫兹波照射时, 有些频率的光波会被物质选择性地吸收, 导致该频率的光强度减弱. 当某频率太赫兹波辐射能量ΔE辐射(式1)与跃迁的能级差ΔE振动(式2)相同时, 会引起分子转动或振动, 同时从基态跃迁到更高能级的激发态, 从而产生吸收谱线, 如式(3), 式(4)[8].
ΔE辐射=hvl,(1)
式中:vl为太赫兹辐射频率.
ΔE振动=ΔV·hv,(2)
式中: ΔV为分子振动量子数差;h为普朗克常量;v为分子振动频率.
产生吸收光谱前提为
ΔE辐射=ΔE振动,(3)
即
vl=vΔV.(4)
因此, 只有当太赫兹辐射频率等于分子振动频率与振动量子数差值的乘积时, 分子才能产生太赫兹吸收光谱, 说明物质太赫兹光谱与分子在太赫兹波段的运动状态之间存在着密切的关系.
1.2 模拟计算理论
密度泛函理论(Density Functional Theory, DFT)是利用电子密度泛函来模拟电子相关, 通过电子密度函数来描述和确定结构性质的理论[9]. H-K第一定理指出体系的基态能量是电子密度的泛函, 并且基于密度泛函理论计算的非经典力场能够精确地描述分子的振动, 因此通过DFT方法可以在低频段对物质进行振动频率计算、 振动模式分析和物质分子结构及相关性质研究. 选择合适的泛函方法是计算相关性质的保证, 在实际计算中, PW91PW91、 BP86、 VSXC、 BLYP和PBE等方法应用较广泛[10]. 在对构型、 能量及光谱性质计算时, 杂化泛函计算结果较准确.
分子建模是模拟计算的基础, 为了避免由分子模型的微小差别引起计算结果的较大误差, 一般选用剑桥大学晶体库CCDC中分子模型. 计算能量对原子位置的一阶导数, 直到该值为零, 则完成对该分子的优化, 继续计算能量对原子位置的二阶导数, 则可得到分子的振动频率.
2 RDX太赫兹吸收光谱模拟计算
黑索金(RDX), 分子式C3H6N6O6, 化学名为环三亚甲基三硝胺, 其分子平面结构如图 1 所示[11].
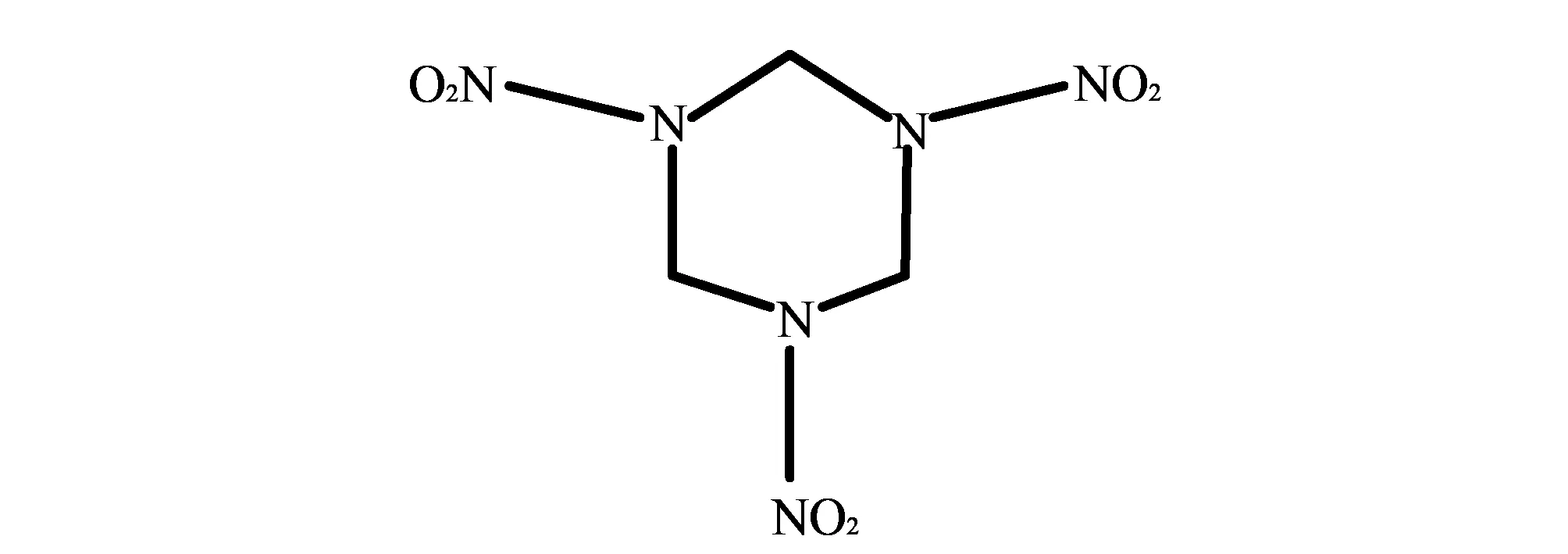
图 1 RDX分子平面结构Fig.1 Plane structure of RDX
为了更加准确地模拟计算RDX的太赫兹光谱, 按照对分子间作用力的考虑程度, 基于密度泛函理论计算两种不同构型RDX的太赫兹光谱. 借助Gaussian 09软件对单分子与二聚体构型进行气态振动模式计算.
2.1 RDX单分子模拟计算
计算方法选择应用最广泛且计算结果较准确的B3LYP杂化泛函, 基组选择为6-31G*[12]. 优化后的RDX如图 2 所示.
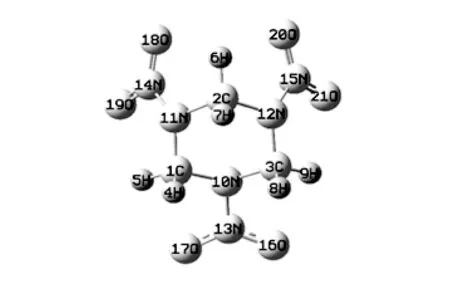
图 2 优化后RDX分子Fig.2 Optimized RDX molecule
优化结构显示由原子13N、 16O、 17O组成的硝基(-NO2)所在平面和三个碳原子所在平面平行, 而由14N、 18O、 19O以及15N、 20O、 21O组成的两个硝基所在平面则和三个碳原子所在平面有一定角度, 并且呈对称分布. 其太赫兹光谱如图 3 所示.
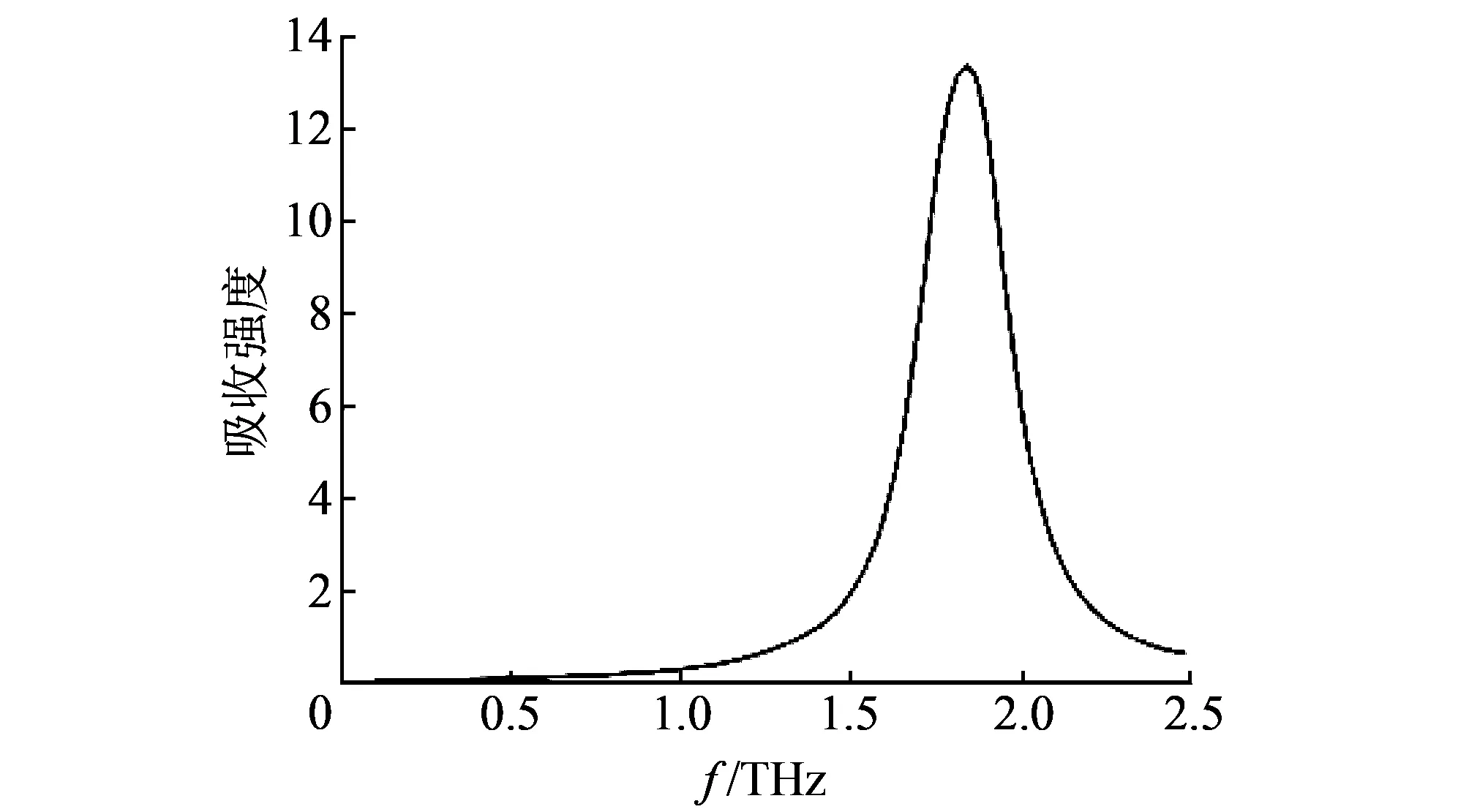
图 3 单分子RDX太赫兹仿真光谱Fig.3 Terahertz simulation spectrum of single RDX molecule
观察图 3 可知, 基于单分子构型的模拟计算只得到了RDX在0.1~2.5 THz频段内1.88 THz 处的一个吸收峰. 因为单分子模型只考虑分子内振动, 忽略了分子间作用、 晶格振动对振动光谱的影响, 从而导致了理论谱中峰位的缺失[13]. 因此, 在模拟计算中采用单分子模型很难精确地反映物质太赫兹光谱全貌.
2.2 RDX二聚体模拟计算
为了克服单分子模型只考虑分子内振动而导致计算结果差异较大的不足, 同时为了探究分子间作用力对吸收光谱的影响, 建立了RDX二聚体构型, 理论方法和基组选择与单分子构型相同. 如图 4 为优化后的RDX二聚体.
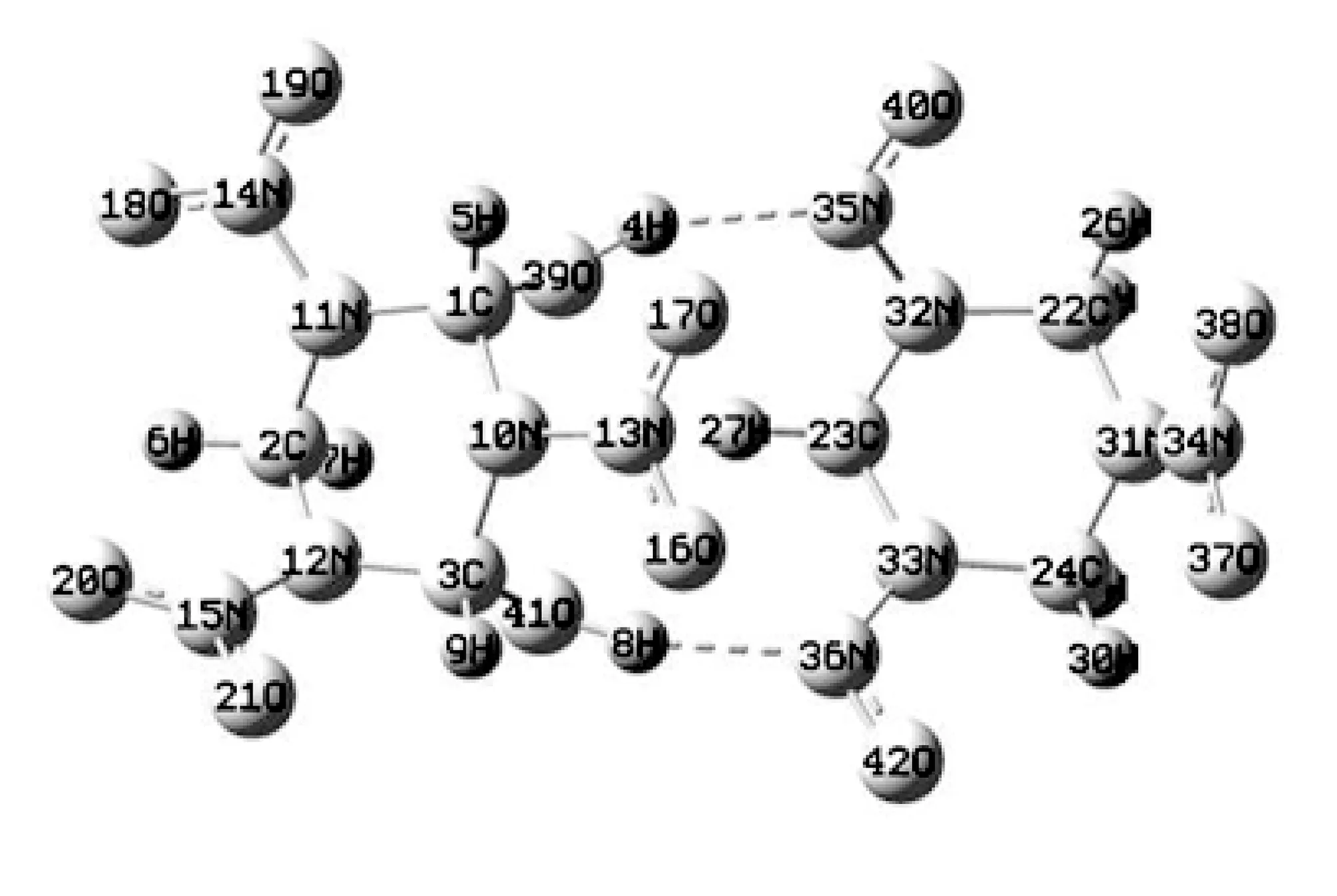
图 4 优化后RDX二聚体Fig.4 Optimized RDX dimer
优化结构显示两个RDX分子通过36N、 41O和8H形成分子间氢键, 键长1.92 nm, 键角157.8°, 以及35N、39O和4H形成分子间氢键, 键长1.95 nm, 键角148.8°, 其太赫兹光谱如图 5 所示.
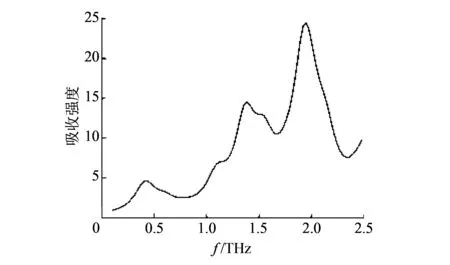
图 5 RDX二聚体太赫兹仿真光谱Fig.5 Terahertz simulation spectrum of RDX dimer
提取图 5 中RDX二聚体太赫兹光谱的吸收峰峰位, 得到特征吸收峰分别为0.37, 1.08, 1.38, 1.58, 1.98 THz.
3 实 验
3.1 系统平台
图 6 为自搭建的THz-TDs系统, 所用激光器为锁模钛宝石激光器, 中心波长800 nm, 稳定测量范围0.1~2.0 THz, 重复频率80 MHz, 脉宽100 fm.

图 6 太赫兹时域光谱系统Fig.6 Terahertz time-domain spectroscopy system
该系统的核心是太赫兹时域光谱技术, 其工作原理如图 7 所示.

图 7 太赫兹时域光谱系统工作原理图Fig.7 Schematic diagram of terahertz time-domain spectroscopy system
该系统主要分两路工作, 一路为泵浦光激发产生太赫兹脉冲后测量待测样品, 一路为探测光经过反射和聚焦后, 与泵浦光在同一时间到达太赫兹光电探测晶体碲化锌上. 碲化锌经过光电转换形成电场, 从而引起探测脉冲的偏振态改变, 这种改变被光电采样系统采集, 并经锁相放大器提高信噪比后, 输入计算进行信号处理和光学参数提取.
3.2 实验结果
提取0.1~2.0 THz范围内的光谱数据, 利用Origin经过Daubechies4小波降噪, 设置Thresholding Level为1, Threshold of every level为50%, 降噪及平滑后的光谱如图 8 所示.
利用Origin进行光谱峰值提取, RDX在0~2 THz 频谱范围内的特征吸收峰为: 0.26, 0.80, 1.05, 1.37, 1.55, 1.93 THz.
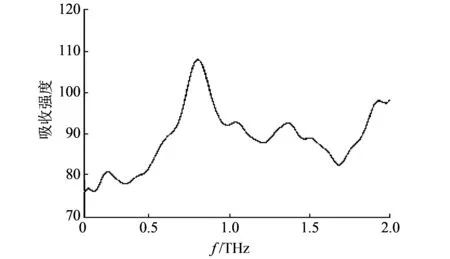
图 8 RDX太赫兹光谱Fig.8 RDX terahertz spectrum
4 分 析
4.1 模拟结果与实验结果对比
利用Gaussian进行计算时, 每种方法计算的频率会与实验结果之间存在一个系统误差, 故计算的频率值会略高于实验值, 需要乘以一个校正因子, 理论方法B3LYP基组6-31G*校正因子为0.961 3[14]. 提取实验与仿真结果中RDX吸收峰对照如表 1. 表中“-”表示未得到相关仿真值.
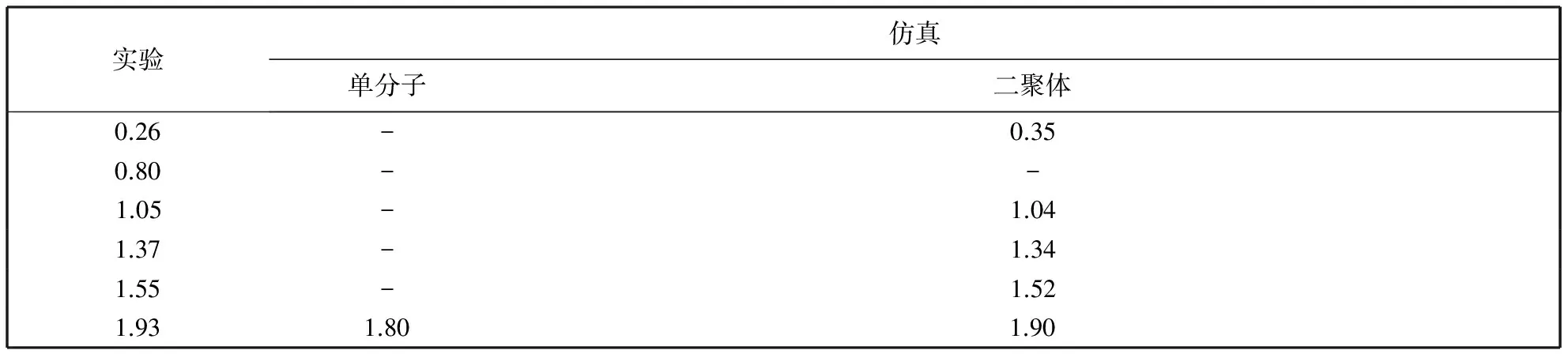
表 1 RDX特征吸收峰实验结果与仿真结果对比
分析表 1 可知, 基于单分子RDX仅仅计算得到1个与实验结果相近的吸收峰, 原因是单分子构型只考虑了分子内作用而忽略了分子间氢键作用对炸药太赫兹光谱的影响. 基于二聚体构型计算结果则在吸收峰个数、 位置以及谱线符合度上都与实验结果吻合地较好, 但是没有得到与0.80 THz 相对应的吸收峰, 可能是由于二聚体模型忽略了晶格振动或声子模式造成的, 各吸收峰峰位也有一定的偏移, 但在可接受范围内, 可能是由于软件计算默认的温度为零度, 而实验中为室温引起的[15].
4.2 特征吸收峰指认
计算得到的单分子1.80 THz和二聚体1.90 THz 与实验结果1.93 THz相对应, 可推断1.93 THz是由分子内振动引起的. 利用Gaussian View演示该处分子振动模式如图 9 所示. 图中箭头的方向表示振动方向, 线的长短表示振动大小. 观察分析可知吸收峰1.93 THz是由与三个碳原子所在平面平行的硝基的伸缩振动以及另外两个硝基围绕各自的N-N键扭动引起的.
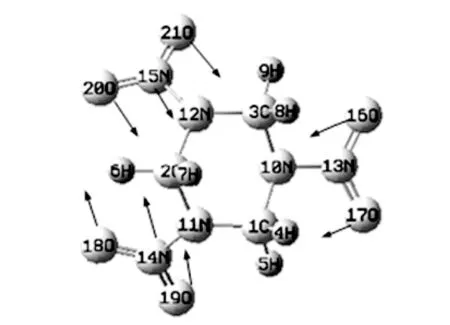
图 9 1.93 THz RDX分子振动模式Fig.9 RDX molecular vibration mode at 1.93 THz
一般, 分子外振动频率比内振动频率低, 因为在分子靠近时分子间会形成氢键. 与共价键和离子键相比, 氢键键能较弱, 且由氢键连接的分子运动质量较大, 因此由分子间氢键作用产生的特征吸收低于分子内共振频率, 处于更低的频率范围内, 且吸收强度也较弱. 对比单分子与二聚体得到的太赫兹光谱, 可以分析得出0.26, 1.05, 1.37, 1.55 THz吸收峰是由分子间氢键作用引起的. 0.80 THz吸收峰在单分子和二聚体模型中都没有出现, 可初步推断该特征吸收峰是由晶体原子在格点附近的热运动(晶格振动)引起的.
5 结 论
本文提出了一种将RDX二聚体作为初始构型的仿真方法. 该方法在对RDX单分子太赫兹吸收光谱仿真的基础上, 对由氢键连接的RDX二聚体进行仿真, 并借助可视化模块分析RDX太赫兹吸收光谱的形成机理, 同时搭建了太赫兹时域光谱系统测得了RDX的吸收光谱.
仿真与实验结果表明:
1) 与单分子模型得到的仿真光谱存在多处峰位缺失相比, 基于RDX二聚体模型的仿真光谱在吸收峰个数、 位置以及谱线符合度上与实验吻合地更好.
2) RDX在太赫兹波段产生特征吸收不仅与分子基团的伸缩振动、 变形振动相关, 也与分子间作用力有密切联系.
3) 太赫兹时域光谱技术和光谱模拟计算相结合的方法, 为研究炸药在太赫兹波段的吸收特征、 产生机理以及结构特性提供了重要的理论基础.
[1] 戴泽林, 许向东, 谷雨. 再生纤维素太赫兹光谱的实验与理论研究[J]. 光谱学与光谱分析, 2017, 37(3): 697-703.
Dai Zeling, Xu Xiangdong, Gu Yu. Experimental and theoretical study on terahertz spectra for regenerated cellulose[J]. Spectroscopyand Spectra Analysis, 2017, 37(3): 697-703. (in Chinese)
[2] 何伟平, 黄菊, 王德堂. 苯乙烯化苯酚的密度泛函研究[J]. 原子与分子物理学报, 2017(5): 839-846.
He Weiping, Huang Ju, Wang Detang. Density functional study of styrenated phenol[J]. Journal of Atomic and Molecular Physics, 2017(5): 839-846. (in Chinese)
[3] Allis D G, Zeitler J A, Taday P F, et al. Theoretical analysis of the solid-state terahertz spectrum of the high explosive RDX[J]. Chemical Physics Letters, 2008, 463(1): 84-89.
[4] 冯瑞姝, 徐栩. RDX在太赫兹波段特征吸收峰成因的研究[J]. 大庆师范学院学报, 2010(6): 78-80.
Feng Ruishu, Xu Xu. The study of the formationof characteristic absorption peaks of the RDX in the terahertz band[J]. Journal of Daqing Normal University, 2010(6): 78-80. (in Chinese)
[5] 闫慧. 有机分子太赫兹光谱的实验与理论计算研究[D]. 西安: 西安光学精密机械研究所, 2012.
[6] 黄玉, 孙萍, 张正. 拉西地平的太赫兹光谱实验研究[J]. 光谱学与光谱分析, 2017, 37(8): 2356-2359.
Huang Yu, Sun Pin, Zhang Zheng. Experimental study on terahertz spectrum of lacidipine[J]. Spectroscopyand Spectra Analysis, 2017, 37(8): 2356-2359. (in Chinese)
[7] Ahmed T, Azad A K, Chellappa R,et al. Vibrational signatures in the THz spectrum of 1,3-DNB: A first-principles and experimental study[J]. EPL (Europhysics Letters), 2016,114(3): 10-15.
[8] Chamorro Posada P, Silva Castro I. A study of the far infrared spectra of N-acetyl-d-glucosamine using THz-TDS, FTIR and semi empirical quantum chemistry methods[J]. Journal of Spectrascopy, 2016, 36: 18-22.
[9] Seyfi S, Alizadeh R, Darvish Ganji, et al. Synthesis, spectral and luminescence study, crystalstructure determination and DFT calculation of bi-nuclear palladium(II) complexes[J]. Spectrochimica Acta Part A: Molecular and Biomolecular Spectra, 2017, 190: 298-311.
[10] 胡银, 宁艳利, 康莹. DNGTz二聚体分子间相互作用的密度泛函理论计算[J]. 火炸药学报, 2017, 40(5): 30-38.
Hu Yin, Ning Yanli, Kang Ying. Intermolecular interactions of DNGTz dimers: a density functional theoretical calculation[J].Chinese Journal of Explosives and Propellants, 2017, 40(5): 30-38. (in Chinese)
[11] 张瑾, 崔洪亮, 施长城, 等. 环氧树脂胶的太赫兹光谱特性研究[J]. 光谱学与光谱分析, 2016, 36(4): 919-923.
Zhang Jin, Cui Hongliang, Shi Changcheng, et al. Terahertz spectroscopic study on the property of epoxy resin adhesive[J]. Spectroscopy and Spectral Analysis, 2016, 36(4): 919-923.
[12] Kumar, Rajesh, et al. Molecular structure,sec-ttroscopic(FT-IR, FT Raman, UV, NMR and THz) investigation and hyperpolarizability studies of 3-(2-Chloro-6-fluorophenyl)-1-(2-thienyl) prop-2-en-1-one[J]. Journal of Molecular Structure, 2017, 11(29): 292-304.
[13] Yang X, Wei D, Yan S, et al. Rapid and la-bel-free detection and assessment of bacteria by terahertz time-domain spectroscopy[J]. Journal of Biophotonics, 2016, 9(10): 1050-1058.
[14] 郭勇. 高斯使用指南[M]. 西安: 西安工业大学出版社, 2005.
[15] 高慧玲. 金属咔咯的量子化学计算和光谱研究[D]. 合肥: 中国科学技术大学, 2013.
猜你喜欢
昆钢科技(2022年2期)2022-07-08
能源工程(2022年1期)2022-03-29
昆钢科技(2021年4期)2021-11-06
雷达学报(2018年1期)2018-04-04
雷达学报(2018年1期)2018-04-04
雷达学报(2018年1期)2018-04-04
分析化学(2018年12期)2018-01-22
科技视界(2016年19期)2017-05-18
山东工业技术(2016年15期)2016-12-01
火炸药学报(2014年3期)2014-03-20
