
氧化锰干法联合同步脱硫脱硝中脱硝反应动力学
2018-01-25 23:36朱超越陈学玺
化学反应工程与工艺 2017年5期
朱超越,陈学玺
青岛科技大学化工学院,山东 青岛 266042
随着国家对大气污染物排放的管控,按照GB 13223-2011要求,SO2和NOx的排放量分别控制到35和50 mg/m3,各地企业单位纷纷安装烟气脱硫脱硝处理装置。脱硫脱硝技术可分为单独脱硫脱硝技术和联合同步脱硫脱硝技术。单独脱硫脱硝技术中,石灰石-石膏法烟气湿式脱硫技术(WFGD)和选择性催化还原(SCR)法脱硝技术应用最为广泛。烟气湿法脱硫技术脱硫率可达95%以上,但该技术副产成分复杂,脱硫石膏成为固体废弃物。SCR法脱硝技术的催化剂起活温度高,也易受SO2毒害,所以不少厂家在对烟气采用湿法脱硫之后,再采用 SCR技术进行脱硝时,必须将烟气重新加热以适应SCR技术要求。因此,采用先脱硫再脱硝的方法,虽然烟气中的NOx和SO2含量可以达到国家规定的排放标准,但是存在巨量的固体废弃物、消耗大量氨以及由此带来的二次污染等缺点,给企业带来沉重经济负担。为降低生产成本,联合同步脱硫脱硝技术应运而生,联合同步脱硫脱硝是基于在同一工艺段中通过氧化锰等复杂化合物催化氧化并同步吸收的方式实现脱硫脱硝一体化脱除。催化氧化技术为治理污染提供了一条高效且经济的新思路,并有望实现“超低排放”[1]。目前国内外都将干法联合脱硫脱硝技术作为重点的研究对象[2-5]。本工作利用烟气中过剩氧气和氧化锰的催化氧化作用,将NO和SO2转化成容易吸收的NO2和SO3,实现烟气中硫和氮氧化物的高效脱除[6-8],探讨了氧化锰固体吸附剂在单独脱硝、先脱硫后脱硝以及联合同步脱硫脱硝过程中脱硝和脱硫的效率,并建立了单独脱硝和联合同步脱硫脱硝得反应动力学方程,为脱硫脱硝干式一体化技术的深入开发和商业化应用提供理论依据。
1 实验部分
1.1 吸收剂的制备
以自制氧化锰作为固体吸收剂进行烟气脱硫脱硝。先将氢氧化钙白色粉末混于水中,再将其缓慢加入到具有相同物质的量的硝酸锰溶液中,使其充分反应生成氢氧化锰悬浮液,经过滤和多次水洗,得到较纯净的氢氧化锰泥状物,放入烘箱中焙烧得到氧化锰吸收剂,记为MnOx(1<x<2)。
1.2 实验装置
吸收剂脱硫脱硝性能实验在固定床吸收实验台上进行,装置如图1所示,系统主要由模拟烟气配气装置、固定床吸附反应器、KM950型烟气分析仪和尾气处理装置4个部分组成。

图1 固定床吸附实验装置Fig.1 Fixed bed adsorption experiment device
设置模拟烟气的各组份(主要成分为 SO2和NOx),实验前先将模拟烟气转至旁路,根据实验要求调节流量计,待数据稳定后,将模拟烟气转至主路,通过烟气分析仪记录实时数据。
1.3 分析方法
采用滴定法定量分析氧化锰,在硫磷混合酸介质中和碘化钾过量的条件下,MnO2可与KI反应,定量析出碘,用硫代硫酸钠标准溶液滴定碘,可计算样品中MnO2的量;紫外分光光度计法定量分析硝酸锰,硝酸根离子中含有-N=O基团,此基团在紫外光区有强烈的吸收,并且其含量与吸光度之间的关系符合朗伯-比尔定律,因此硝酸根离子的检测可以利用紫外分光光度计来进行检测;离子色谱法定量分析硫酸锰,采用阴离子交换柱为分离柱,用氢氧化钾淋洗液发生器产生的15×10-3mol/L的KOH溶液为流动相,流量为1.0 mL/min,柱温为30 ℃条件下进行色谱分离和测定[9]。
2 动力学模型建立
本实验中固体吸收剂的脱硫脱硝反应可用以下化学方程式表示:
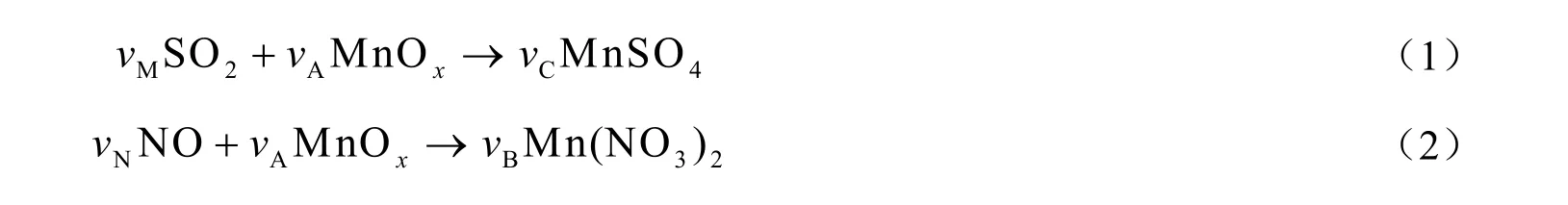
每次取5 g氧化锰吸收剂放置于固定床反应器内,反应体系可视为微分反应器。
以单位时间内锰原子质量变化的百分数来表示反应速率,则该反应的瞬时速率(Ri,min-1)可用氧化锰的瞬时消耗速率表示,也可以用硝酸锰或者硫酸锰的瞬时生成速率来表示,见式(3)。

式中:mi为生成的硝酸锰或硫酸锰的量,mol;m0为固体样品中含氧化锰的量;vi分别为氧化锰、硝酸锰和硫酸锰的化学计量系数;t为时间,min。
当固体单独脱硫或脱硝生成硫酸锰或者硝酸锰时,从 0~t时间间隔内对瞬时反应速率积分后,再对时间取平均即可得到平均反应速率(Rav,min-1)。
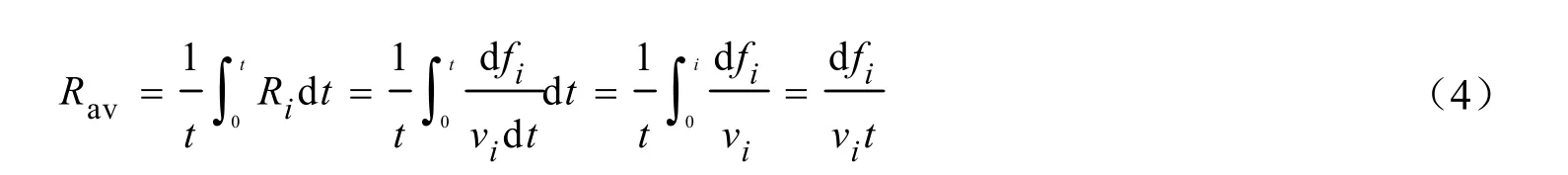
式中:fi为固体吸收剂中氧化锰、硝酸锰以及硫酸锰的物质的量分数,其分别为fA=mA/m0,fB=mB/m0,fC=mC/m0。
先前实验证实氮气、氧气和二氧化碳气体的存在不会对NO和SO2吸收反应产生影响。反应过程中的变量是混合气中NO的分压(PNO)、温度(T)和固体吸收剂氧化锰表面有效空位率(θA)。
在吸收反应过程中,吸收剂氧化锰固体表面的有效位置被NO和SO2吸附并占据后,反应生成硫酸锰和硝酸锰,因此氧化锰表面的空位率与吸收剂中的氧化锰、硝酸锰和硫酸锰含量建立了关系[10,11]。
3 结果与讨论
3.1 反应条件对脱硝效率的影响
3.1.1 NO分压的影响
在氧气的体积分数为6%,恒温120 ℃的条件下,考察不同NO气体分压对氧化锰的脱硝效果的影响,结果如图2所示。由图可知,随着NO气体分压的增加,Mn(NO3)2生成率不断增加。这是由于反应物浓度增加,化学反应速率加快,吸收剂瞬时脱硝速率得以提高[12,13]。
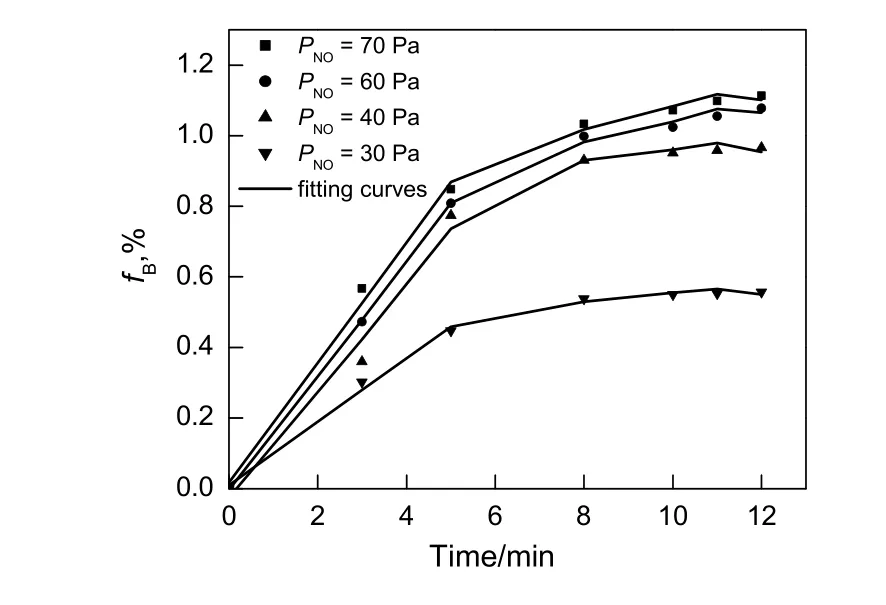
图2 NO分压对Mn(NO3)2生成率的影响Fig.2 Effect of NO pressure on production rate of Mn(NO3)2
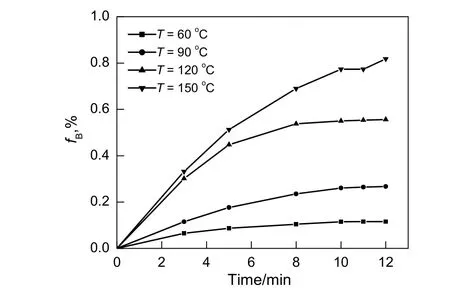
图3 温度对Mn(NO3)2生成率的影响Fig.3 Effect of temperature on production rate of Mn(NO3)2
3.1.2 温度的影响
在氧气的体积分数为6%,NO气体分压为30 Pa的条件下,考察反应温度对脱硝实验的影响,结果如图3所示。由图可知,在NO分压一定的情况下,固体吸收剂瞬时脱硝速率随着温度的升高而增大[14],并在短时间内没有下降的趋势,这说明反应温度的升高,能够显著提高化学反应速率。因此该温度范围是氧化锰脱硝的合适操作范围。
3.2 内、外扩散的消除
气固反应外扩散阻力存在于相界面,一般选用足够大的空间速度进行动力学实验,在较细粒度和一定温度下,保持空速不变,逐渐增加反应物装量,反应物转换率基本不随之变化,可认为外扩散对反应的影响基本消除。在空速为1 000 h-1,氧化锰粒度最大为1 µm,分别考察在恒压不同温度及恒温不同压力下反应物装量对转化率的影响,结果如图4所示。由图可知,反应物装量的增加几乎不影响Mn(NO3)2生成率,说明在本实验条件下外扩散对反应的影响已消除。
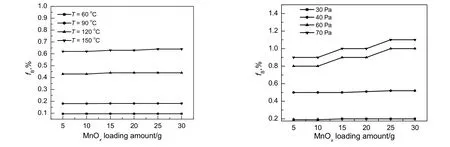
图4 MnOx装量对Mn(NO3)2生成率的影响Fig.4 Effect of MnOx loading amount on production rate of Mn(NO3)2
内扩散对反应的影响可通过Weisz-Prater判据进行判定。

式中:CWP为Weisz-Prater参数;rA为反应速率,min-1;r为颗粒半径,m;De为有效扩散系数,m2/s;CAS为反应物初始浓度,mol/L。
当CWP远小于1时,反应过程不受内扩散影响,当CWP远大于1时,反应过程为强内扩散控制。本实验所用的氧化锰粒径较小,最大粒径为1 μm,密度为450 kg/m3,通过估算CWP远远小于1,可以忽略内扩散对反应的影响。
3.3 动力学参数求取
MnOx颗粒小于1 μm,生成Mn(NO3)2固体,忽略扩散传质阻力的情况下,反应动力学速率方程为[15-17]:

式中:Rav1为固体吸收剂单独脱硝的平均反应速率,min-1;ks为吸收反应的反应速率常数,min-1;n为吸收反应针对PNO的反应级数;θA为氧化锰表面空隙率,%。
根据Arrhenius方程:

式中:A是指前因子,min-1;E是吸收反应的活化能,kJ/mol;R是理想气体常数,8.314 J/(mol·K)。
在温度恒定为120 ℃的情况下,NO的分压从30 Pa到70 Pa,由固体中硝酸锰含量与时间的关系求得平均反应速率,并将lnRav1对lnPNO作图,结果如图 5所示。经计算得图中直线斜率为 0.8,故动力学方程中n为0.8。
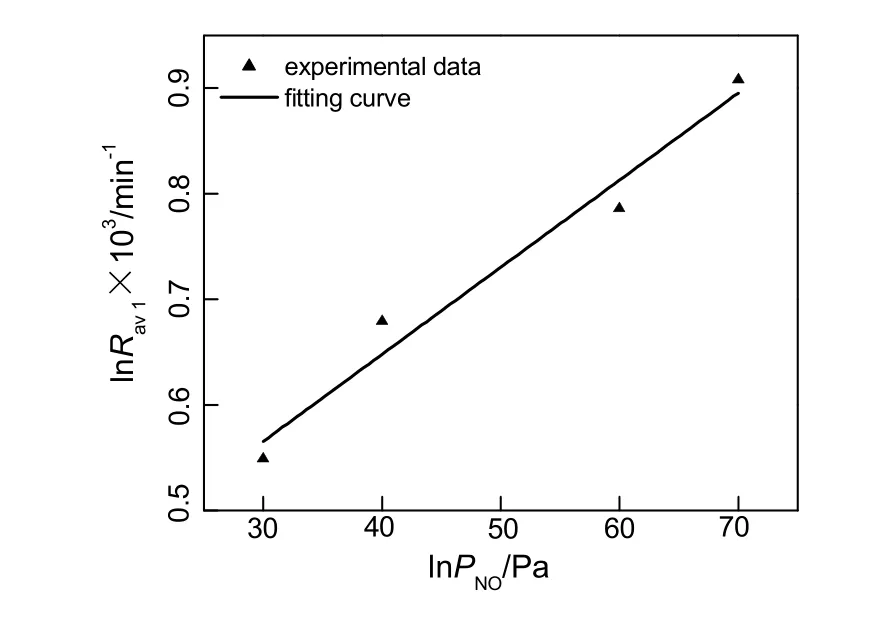
图5 平均反应速率与PNO的关系Fig.5 The relationship between the average reaction rate and PNO
在固体吸收剂与气体进行吸收反应时,吸收剂颗粒表面的活性中心逐渐被NO占据,θA下降,满足θA=1-θB关系,θB是活性中心被 NO 气体分子占据的覆盖率,它正比于fB,反应过程中θA可以用1-fB表示。当PNO为30 Pa时,温度从60 ℃变化到150 ℃,根据图3不同温度下Mn(NO3)2生成率随时间的变化情况,求得各温度下固体吸收剂脱硝的平均反应速率与温度的关系。将lnRav1对T-1作图,结果见图6。根据式(8),由图6直线的斜率和截距可计算得到该吸收反应的活化能为24.16 kJ/mol,指前因子为464.64 min-1。
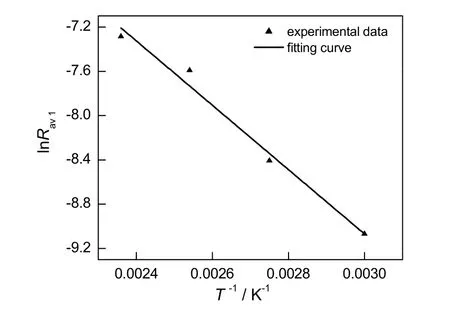
图6 Rav1与T -1的关系Fig.6 The relationship between the Rav1 and T -1
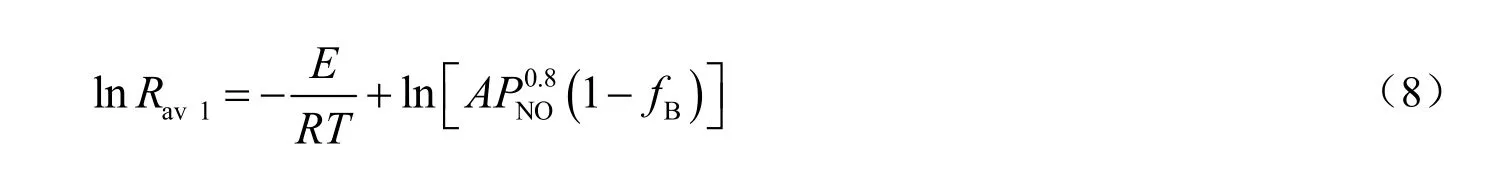
固体吸收剂单独脱硝时,反应速率可用下式表示:

3.4 脱硫后再脱硝的结果
在固体吸收剂单独脱硫完成后,将原料气改换为N2,O2和NO的混合气,在NO的压力为70 Pa,SO2压力为40 Pa条件下,继续在120 ℃下进行固体吸收剂的脱硝实验,与之前用新鲜固体吸收剂颗粒直接脱硝进行对比。结果如图7所示。从图可知,与直接脱硝相比,脱硫后吸收剂的脱硝效果下降近50%,这是由于更易吸收的SO2气体分子反应占据一部分活性位置,给脱硝过程增加了一定的吸收难度,因此造成了脱硝效率的下降。
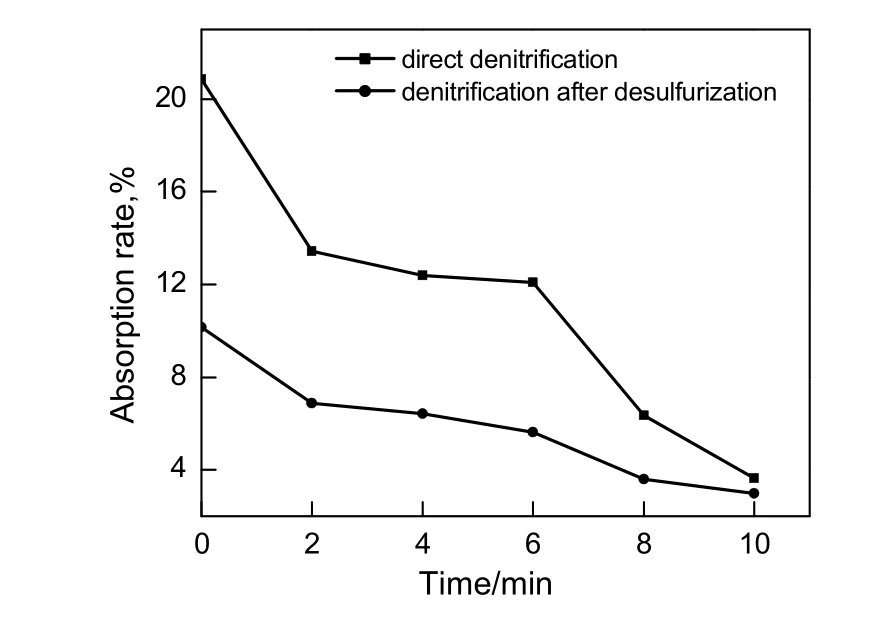
图7 直接脱硝与先脱硫后脱硝吸收率比较Fig.7 Comparison of absorption rate between the direct denitrification process and denitrification after desulfurization process
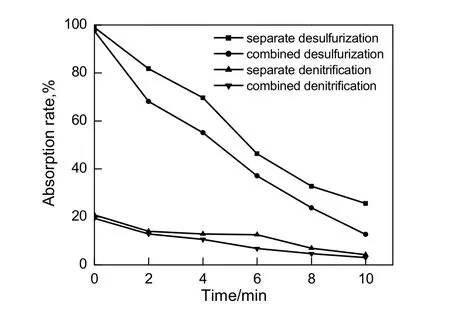
图8 脱硫脱硝吸收率在单独进行和联合进行时的比较Fig.8 Comparison of desulfurization and denitrification absorption rates between direct and simultaneous denitrification and desulfurization process and combined process
3.5 联合同步脱硫脱硝的实验结果
先脱硫后脱硝的实验,已明确显示出脱硫和脱硝时,两个过程会相互影响,产生活性中心的竞争。为探讨联合同步脱硫脱硝反应结果,选择反应温度120 ℃,NO的压力为70 Pa,SO2压力为40 Pa时进行联合同步脱硫脱硝实验,并对单独脱硝过程的反应动力学方程进行简单修正。
由图8可知,单独脱硫与联合脱硫的吸收率随时间变化逐渐下降,单独脱硫与联合脱硫相比,其吸收率要高,单独脱硫吸收率的下降速率慢于联合脱硫吸收率的下降速率,单独脱硫吸收率的下降速率约为联合脱硫吸收率的 0.93倍;而对于单独脱硝与联合脱硝过程来说,两者的差距不大,相同反应时间下最大的吸收率差值约为2.23%,单独脱硝吸收率的下降速率约为联合脱硝吸收率下降速率的0.95倍。
为建立单独脱硝和联合脱硝两个过程反应动力学方程的联系,引入修正因子,对单独脱硝反应动力学方程进行一定程度的修正,即可得到近似的联合同步脱硫脱硝过程中脱硝的平均反应速率表达式,如式(10)。

其中,ε为修正因子,其值为0.95。可先单独脱硝测得相关的动力学数据,再用此修正式来近似表示联合同步脱硫脱硝过程中脱硝反应的平均反应速率[18]。
3.6 动力学模型检验
为了验证求得的动力学模型的可靠性,分别在60 ℃,30 Pa和150 ℃,70 Pa下,将固体吸收剂单独脱硝和联合同步脱硫脱硝中脱硝实验结果和动力学模型计算结果进行比较,结果见图 9和 10。由图可知,在本实验条件下,由动力学模型计算所得的Mn(NO3)2生成率与实验值相近,经计算得其相对误差均小于6%,说明所求得的动力学模型预测的结果与实验结果吻合良好。
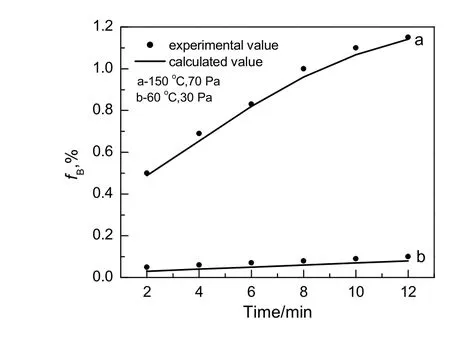
图9 单独脱硝实验结果与计算值比较Fig.9 Comparison of direct denitrification experimental value and calculated value

图10 联合同步脱硫脱硝中脱硝实验结果与计算值比较Fig.10 Comparison of denitrification experimental value and calculated value for simultaneous desulfurization and denitrification
4 结 论
a)在单独脱硝中,MnOx脱硝反应速率随温度升高、NO气体分压的增加而加快。
b)先脱硫再脱硝,脱硝效率下降近一半。在联合同步脱硫脱硝的过程中,脱硫与脱硝相互影响但对脱硝效率影响较小,联合同步脱硝效率只比单独脱硝少5%,因此,联合同步脱硫脱硝MnOx使用效率更高。
c)将单独脱硝反应动力学方程进行简单修正,得到联合同步脱硫脱硝过程中脱硝的反应速率动力学方程,反应级数为0.8,反应的活化能为24.16 kJ/mol,指前因子为464.64 min-1,修正参数为0.95。该动力学模型预测的结果与实验结果吻合良好。
[1]方宁杰. 烟气脱硫脱硝技术的现状和发展趋势[J]. 四川化工, 2016, 19(5): 29-32.Fang Ningjie. Present situation and trends of flue gas desulfurization and denitrifcation technology[J]. Sichuan Chemical Industry, 2016,19(5): 29-32.
[2]朱金伟, 张 凡, 王洪昌. 燃煤烟气脱硫脱硝技术的发展趋势[J]. 环境工程技术学报, 2015, 5(3): 200-204.Zhu Jinwei, Zhang fan, Wang Hongchang. Analysis on development trend of desulfurization and denitration technologies for coal-fired flue gas[J]. Journal of Environmental Engineering Technology, 2015, 5(3): 200-204.
[3]曹会龙. O2/CO2气氛下改性方解石煅烧分解与固硫特性研究[D]. 武汉: 华中科技大学, 2011.
[4]马 强. 烟气中多种污染物超低排放的活性分子氧化及一体化脱除机理研究[D]. 杭州: 浙江大学, 2016.
[5]杨东月. 燃煤电厂烟气综合净化技术研究[D]. 北京: 华北电力大学, 2015.
[6]陈学玺, 韩香莲, 朱超越, 等. 一种以氧化锰为循环吸收介质的烟道气脱硝方法: CN105771652A[P]. 2016-07-20.
[7]陈学玺, 陈春光, 朱超越, 等. 一种以氢氧化锰为循环工作介质的烟道气深度脱硫脱硝干式一体化方法: CN106861410A[P].2017-06-20.
[8]陈学玺, 陈春光, 韩香莲, 等. 一种完全脱除烟道气中SO2联产石膏晶须的方法: CN105854569A[P]. 2016-08-17.
[9]夏玉宇. 化验员实用手册[M]. 北京: 化学工业出版社, 2012: 561-562, 1055-1067.
[10]Liang H, Shingohara K, Minoshima H. Analysis of constant rate period of spray drying of slurry[J]. Chemical Engineering Science,2001, 56(6): 2205-2213.
[11]Yang H M, Kim S S. Experimental study on the spay characteristics in the spray drying absorber[J]. Environ Sci Technol, 2000, 34(21):4582-4586.
[12]Blumrich S, Engler B. DESONOX/REDOX-process for flue gas cleaning: a flue gas purification. process for the simultaneous removal of NOx and SO2resp CO and UHC[J]. Catalysis Today, 1993, 17(1/2): 301-310.
[13]Clements J S, Mizuno A, Finney W C. Combined removal of SO2, NOx, and fly ash from simulated flue gas using pulsed streamer corona[J]. IEEE Transactions on Industry Applications, 1989, 25(1): 62-69.
[14]Gabriele C, Slglinda P. Efficient simultaneous dry removal of SO2and NOxfrom flue gas over copper-based catalytic materials[J].Developments in Chemical Engineering and Mineral Processing, 2000, 8(5/6): 441-463.
[15]李绍芬. 反应工程[M]. 北京: 化学工业出版社, 2000: 64-69.
[16]陈甘棠. 化学反应工程[M]. 北京: 化学工业出版社, 1997: 54-58.
[17]陈家镛, 夏光祥. 氧化铜与二氧化硫及氧作用生成硫酸铜的动力学[J]. 化工学报, 1965, 1: 1-12.Chen Jiayong, Xia guangxiang. The kinetics of CuO reacting with gasous SO2and O2[J]. Journal of Chemical Industry, 1965, 1: 1-12.
[18]Klang K D, Kun Li, Rothfus R R. Kinetic studies of sulfur dloxide absorption by manganese dioxide[J]. 1976, (10): 886-894.
猜你喜欢
化工管理(2022年13期)2022-12-02
热力发电(2022年5期)2022-06-09
能源工程(2021年5期)2021-11-20
山西冶金(2021年3期)2021-07-27
理化检验-化学分册(2020年5期)2020-06-15
饮食科学(2016年3期)2016-07-04
饮食科学(2016年3期)2016-07-04
现代工业经济和信息化(2016年8期)2016-05-17
中国资源综合利用(2016年3期)2016-01-22
中国资源综合利用(2016年2期)2016-01-22
