
22例儿童中枢神经系统AT/RT临床分析
2018-01-24 08:16:27张晓红牛会林徐涛石军徐令江华
新医学 2018年1期
张晓红 牛会林 徐涛 石军 徐令 江华
非典型畸胎瘤样/横纹肌样瘤(AT/RT)是发生在儿童中枢神经系统的少见的具有极大侵袭性的恶性肿瘤,在儿童中枢神经系统肿瘤中发病率极低,预后差。为探讨其临床特征,笔者将我院诊治的22例中枢神经系统AT/RT患儿的临床资料进行总结分析,以供同行对本病参考。
对象与方法
一、研究对象
2011年1月至2017年7月在我院神经外科和血液肿瘤科住院并经手术病理检查确诊为AT/RT的22例患儿为研究对象。22例中男12例、女10例,发病年龄均小于6岁。
二、方 法
收集所有患儿的临床资料进行总结,分析其特征性临床症状以及影像学改变、病理检查等特点。
结 果
一、一般情况
22例均<6岁,年龄为2个月~5岁。临床症状以呕吐、行走不稳为主。所有患儿发病至诊断时间均超过半个月。22例中有19例原发部位为颅内,1例累及肾脏、3例累及脊髓;3例原发部位为脊髓。22例中12例发生并发症,以梗阻性脑积水为主,见表1。
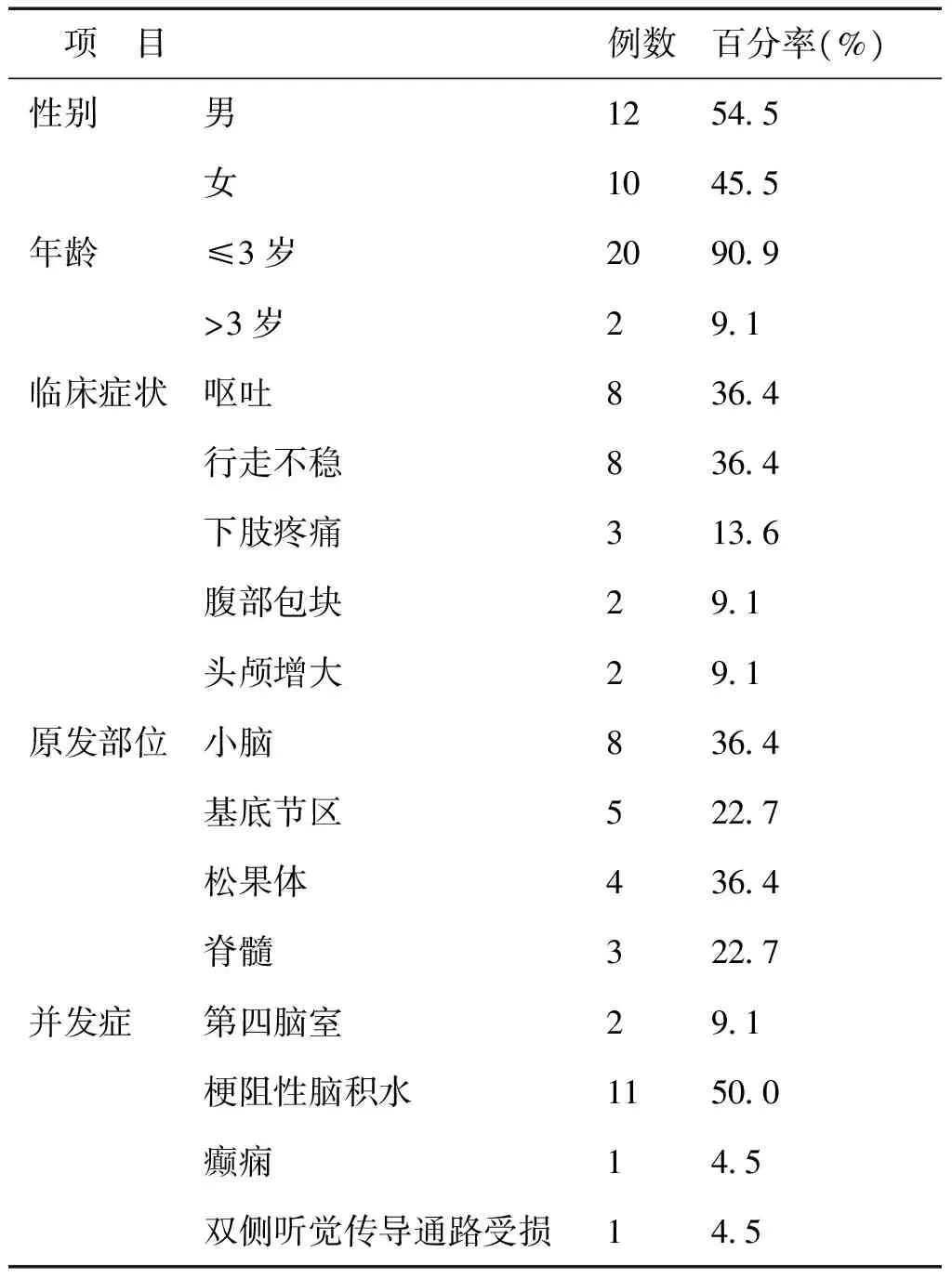
表1 22例AT/RT患儿的一般情况
二、影像学检查结果
22例中16例(72.7%)行头颅MRI平扫+增强检查。5例(22.7%)行CT平扫+增强检查。16例MRI均显示占位性病变,均挤压周围组织造成压迫、梗阻,其中7例(43.8%)为囊实性病变、囊腔大小不一,9例(56.3%)为实性病变;16例MRI的T2加权像均显示为高信号,T1加权像显示为高、等或稍低信号,增强后肿块呈不均匀强化,见图1A~C。22例CT均见不规则软组织密度肿块,增强后呈不均匀强化,见图1D~F。
三、病理检查结果
1.巨检结果
22例手术切除的组织均为灰白或灰红碎组织,均无包膜,大小0.8 cm×0.8 cm×0.1 cm~12.5 cm×11 cm×11 cm,质地中等,切面灰白,灶性淡黄。
2.镜检结果
22例手术切除组织镜检均可见典型的横纹肌样细胞,其中9例(40.1%)有不同程度的坏死,见图1G。10例(45.5%)组织形态多样,表现为肿瘤细胞弥漫成片排列,从状血管增生,可见梭形细胞及透明细胞区域,亦可见腺样结构、菊形团及假菊形团形成,见图1H。12例(54.5%)组织形态单一,表现为肿瘤细胞弥漫成片分布,部分围绕血管排列,肿瘤细胞形态大小较一致,肿瘤细胞边界清楚,核圆形,可见核仁,部分偏位,胞浆红染,呈横纹肌样,可见大量病理性核分裂,凋亡小体易见,见图1I。
3.免疫组织化学检查(免疫组化)结果
22例波形蛋白均为阳性。11例(50%)S-100蛋白阳性,7例(31.8%)淋巴内皮细胞标记D2-40灶性阳性,6例(27.3%)上皮膜抗原阳性,5例(22.7%)CD99阳性,4例(18.2%)神经特异性烯醇化酶阳性,3例(13.6%)抑癌基因P53蛋白阳性,2例(9.1%)CD56阳性,2例(9.1%)平滑肌肌动蛋白阳性, 1例(4.5%)嗜铬素A、神经纤维丝蛋白弱阳性,1例(4.5%)WT1蛋白弱阳性,1例(4.5%)CD34血管内皮阳性。22例神经胶质酸性蛋白、突触素、肌间线蛋白网络结蛋白,角蛋白阴性,21例(95.5%)InI-1/ hsNF5蛋白阴性,3例(13.6%)胎盘碱性磷酸酶(PLAP)阴性。22例的增殖指数Ki-67为10%~80%。
四、治疗及预后
所有病例入院后均行显微微创手术切除肿瘤。21例于病理诊断确定后放弃继续治疗,存活时间1~14个月。1例原发部位在脊髓的患儿,病灶在椎旁,侵及椎管内(T11-L1),临床分期为Ⅲ期,初次住院行肿瘤穿刺活组织病理检查确诊后,交替接受VAC(顺铂+环磷酰胺+吡柔比星)及ICE(异环磷酰胺+依托泊苷+长春地辛)化学治疗,21 d为一疗程,每2个疗程行CT或MRI评估疗效,于化学治疗第4疗程后行显微微创手术切除肿瘤,术后继续交替接受VAC和ICE治疗,总疗程12个,化学治疗间隔期间曾接受病灶放射治疗,总剂量36 Gy,完成治疗5个月后因肿瘤原位复发而放弃继续治疗,随后失访。
A:MRI的T2加权像;B:MRI的T1加权像;C:液体衰减反转恢复(FLAIR)序列;D:CT平扫;E~F:CT增强扫描;G:病理检查显示组织形态单一(苏木素-伊红染色,×100);H:病理检查显示组织形态多样化(苏木素-伊红染色,×100);I:典型的横纹肌样细胞(苏木素-伊红染色,×400)
讨 论
1978年Beckwith[1]等首次报道了中枢神经系统原发性横纹肌样瘤。1996年学者们正式定义该疾病病名为AT/RT[2]。2000年WHO在中枢神经系统肿瘤分类中将此肿瘤归为一种新的神经系统胚胎性肿瘤[3]。
AT/RT临床少见,好发于2岁以下婴幼儿,90%患儿小于5岁,发病中位年龄为26个月[4]。发病率在6~17岁的仅2%~3%,成人少见。AT/RT患者的男女比例为1.6∶1。本研究中的22例均小于6岁,男女比例为6∶5,这与国外同类报道相似[4]。
大部分AT/RA患者的原发部位为中枢神经系统,可发生在颅内任何部位,约60%位于后颅窝,小脑、桥小脑角和脑干多发,而且由于其极具侵袭性,13.6%病例会出现多部位受累[5-7]。本研究中的22例有19例原发部位为颅内,4例累及其它部位,原发部位及多部位发病的比例与其它文献报道相当[5-7]。22例患儿的发病至诊断时间均超过半个月,提示家长重视程度不足, 中枢神经系统AT/RT多以神经系统症状就诊,婴幼儿患儿一般表现为嗜睡、呕吐等非特异性症状,常被家长忽略,另外患儿也可出现发育异常如头颅增大、歪颅、站立异常、行走不稳等症状[7]。
AT/RT的影像学表现虽无诊断特异性,但在定位和鉴别诊断方面具有重要价值[8]。AT/RT容易经脑脊液播散转移,脑膜的异常强化提示肿瘤可能播散,因此MRI检查更有助于协助诊断肿瘤的状态。
确诊AT/RT需通过病理检查。AT/RT组织学形态除了特征性的横纹肌样细胞外,还可有原始外胚肿瘤样区域,肿瘤性上皮或间质等多种异源性构成。本研究中有10例组织形态多样、12例组织形态单一,但均具有特征性横纹肌样细胞。多种细胞成分的存在增加了AT/RT的诊断难度,因此免疫组化具有重要的诊断意义[9]。AT/RT的免疫组化显示波形蛋白为特征性阳性,本研究中的22例即如此,另外22例的增殖指数Ki-67为10%~80%,提示肿瘤细胞增殖能力强,侵袭性高。AT/RT常被认为起源于原始胚胎组织,但具体组织起源未明。近年来其分子遗传学研究有极大进展,InI-1蛋白抗体是鉴别AT/RT与其它组织类型儿童脑肿瘤的敏感特异方法,该指标值得引起重视[10-13]。
AT/RT预后极差,患者常于发病1年内死亡,<3岁患儿对化学治疗反应差,对于<2岁患儿不推荐使用放射治疗,故3岁以内患儿的长期生存机会极低,但对于年长患儿,于接受手术后再接受放射治疗及化学治疗,则有望能长期无病存活[14]。
[1] Beckwith JB, Palmar NF. Histopathology and prognosis of Wilms tumors:results from the first National Wilms tumor Study. Cancer,1978,41:1937-1948.
[2] Biswas A, Kashyap L, Kakkar A, Sarkar C, Julka PK. Atypical teratoid/rhabdoid tumors: challenges and search for solutions. Cancer Manag Res, 2016,8:115-125.
[3] Tanizaki Y,Oka H,Utsuki S,Shimizu S, Suzuki S, Fujii K.Atypical teratoid/rhabdoid tumor arising from the spinal cord-case report and review of the literature.Clin Neuropathol, 2006,25(2):81-85.
[4] Ostrom QT, Chen Y, M de Blank P, Ondracek A, Farah P, Gittleman H, Wolinsky Y, Kruchko C, Cohen ML, Brat DJ, Barnholtz-Sloan JS. The descriptive epidemiology of atypical teratoid/rhabdoid tumors in the United States, 2001-2010. Neuro Oncol,2014,16(10):1392-1399.
[5] Athale UH, Duckworth J, Odame I, Barr R. Childhood atypical teratoid rhabdoid tumor of the central nervous system: a meta-analysis of observational studies.J Pediatr Hematol Oncol,2009,31(9):651-663.
[6] 施伟, 李昊, 赵瑞. 婴幼儿中枢神经系统非典型畸胎样/横纹肌样瘤的治疗体会. 中华神经外科杂志,2016,32(4):349-352.
[7] Li F, Gui Q, Piao Y. Primary supratentorial atypical teratoid/rhabdoid tumor in children: a report of two cases. Child Neurol, 2013, 28(3):399-403.
[8] Warmuth-Metz M, Bison B, Dannemann-Stern E, Kortmann R, Rutkowski S, Pietsch T. CT and MR imaging in atypical teratoid/rhabdoid tumors of the central nervous system. Neuroradiology, 2008, 50(5):447-452.
[9] Al-Hussaini M, Dissi N, Souki C, Amayiri N. Atypical teratoid/ rhabdoid tumor, an immunohistochemical study of potential diagnostic and prognostic markers. Neuropathology, 2016,36(1):17.
[10] Grupenmacher AT, Halpern AL, Bonaldo Mde F, Huang CC, Hamm CA, de Andrade A, Tomita T, Sredni ST. Study of the gene expression and microRNA expression profiles of malignant rhabdoid tumors originated in the brain (AT/RT) and in the kidney (RTK). Childs Nerv Syst, 2013,29(11):1977-1983.
[11] JHoell JI, Gombert M, Bartenhagen C, Ginzel S, Husemann P, Felsberg J, Reifenberger G, Eggert A, Dugas M, Schönberger S, Borkhardt A, Fischer U. Whole-genome paired-end analysis confirms remarkable genomic stability of atypical teratoid/rhabdoid tumors. Genes Chromosomes Cancer,2013,52(10):983-985.
[12] Chakravadhanula M, Tembe W, Legendre C, Carpentieri D, Liang WS, Bussey KJ, Carpten J, Berens ME, Bhardwaj RD. Detection of an atypical teratoid rhabdoid brain tumor gene deletion in circulating blood using next-generation sequencing. J Child Neurol,2014,29(9):81-85.
[13] Zin A, Bertorelle R, Dall'Igna P, Manzitti C, Gambini C, Bisogno G, Rosolen A, Alaggio R. Epithelioid rhabdomyosarcoma: a clinicopathologic and molecular study. Am J Surg Pathol, 2014,38(2):273-278.
[14] Verma V, Johnson CP, Bennion NR, Bhirud AR, Li S, McComb RD, Lin C. Atypical teratoid rhabdoid tumor: long-term survival after chemoradiotherapy. Childs Nerv Syst,2015,31(8):1393-1399.
猜你喜欢
红蜻蜓·低年级(2022年5期)2022-05-25 13:27:34
红蜻蜓·低年级(2022年5期)2022-05-11 21:50:07
中华养生保健(2020年2期)2020-11-16 00:49:30
新医学(2019年6期)2019-07-06 11:30:39
中国医学影像学杂志(2018年9期)2018-10-17 01:27:04
中国自行车(2018年8期)2018-09-26 06:53:34
华人时刊(2018年23期)2018-03-21 06:26:26
结核与肺部疾病杂志(2015年4期)2015-07-18 11:08:21
中国药业(2014年12期)2014-06-06 02:17:24
重庆医学(2014年35期)2014-03-04 08:49:38
