
西安地区夏季臭氧的模拟研究
2018-01-15 10:32:49贝耐芳吴佳睿李国辉
地球环境学报 2017年6期
贝耐芳,冯 添,吴佳睿,,李国辉,
1.西安交通大学 人居环境与建筑工程学院, 西安 710054
2.中国科学院地球环境研究所 黄土与第四纪地质国家重点实验室,西安 710061 3.中国科学院气溶胶化学与物理重点实验室, 西安 710061
臭氧(O3)是大气化学过程中的重要物质。O3在大气平流层和对流层光化学反应中起着主要作用(Seinfeld and Pandis,2006)。在平流层,O3吸收了大部分太阳紫外辐射,为地表生物提供了天然保护伞。在对流层,O3与其光化学衍生物氢氧自由基(OH)是大气中绝大多数还原性气体的关键氧化剂(Brasseur et al,1999)。O3还能吸收大气红外辐射,对气候变化产生影响,是重要的短寿命气候污染物。另外,地表高浓度O3对生态系统和人体健康也有很大的损害(Zhou et al,2011;Cao et al,2012;Verstraeten et al,2015)。因此,O3被很多国家的环境部门,如美国环保署(US EPA)和中国环保部(China MEP),列为关键大气污染物之一。
目前,人们对近地面O3的来源与生成过程有了更加深入的认识,对流层O3的生成机制也逐渐被厘清。对流层O3作为二次污染物,主要由CO、挥发性有机物(VOCs)和氮氧化物(NOx)等前体物通过大气化学、光化学反应生 成(Seinfeld and Pandis,2006;Doherty,2015),生命期大约为22天(Stevenson et al,2006),主要通过进一步参与化学反应、干沉降等方式清除。O3前体物大多来自人为排放,如工业生产、火力发电、居民生活和机动车排放,高排放量集中在人口密集的地区。
我国对O3污染的研究随着我国改革开放进程的深化,工业化和城市化进程的不断加快以及伴随而来的空气污染(De Smedt et al,2010;Lu et al,2011)也在不断推进。目前很多地区针对O3污染开展了诸多研究工作,尤其在京津冀(Wang et al,2006;Lin et al,2008;Tang et al,2009;Xu et al,2011)、长三角(Geng et al,2009;Tie et al,2009;Geng et al,2011;Tie et al,2013)、珠 三 角(Zhang et al,2008;Wang et al,2009;Cheng et al,2010;Wang et al,2011;Li et al,2013)和关中盆地(Shen et al,2010;Wang et al,2012;Xue et al,2017)等地区,并取得了一系列成果。Wang et al(2006)在北京的烟羽中观测到了很高的 O3产量,O3峰值浓度高达 286 nL · L−1。Tie et al(2009)发现上海地区不利的气象条件能够使近地面 O3浓度超过 100 nL · L−1。Wang et al(2009)分析了我国香港地区1994 — 2007年背景大气的地面O3浓度,发现O3浓度呈波动上升趋势,Li et al(2015)对京津冀地区近地面O3形成过程中VOCs所起的作用进行了深入分析。研究指出,烯烃和芳香烃对O3生成潜势(Ozone formation potential,OFP)贡献最大,乙烯(ethene)、间二甲苯(m-xylene)和对二甲苯(p-xylene)、甲苯(toluene)、丙烯(propene)、邻二甲苯(o-xylene)、1-丁烯(1-butene)在O3生成过程中有重要作用,此外机动车排放的VOCs对环境VOCs贡献非常大。我国华北地区的一些研究表明京津冀地区的O3生成过程对VOCs敏感(Wang et al,2006;Tang et al,2012)。Xue et al(2014)指出上海和广州地区O3的生成过程也对VOCs敏感,但兰州地区对NOx敏感。在珠三角地区近地面O3形成的研究中,Li et al(2013)指出珠三角地区O3的生成主要对VOCs敏感,在降低NOx排放的情况下,O3峰值浓度有所降低,但平均浓度反而有所升高。在关中地区的臭氧研究中,西安市O3观测研究发现冬、夏季平均浓度之差可达十倍以上(Wang et al,2012),O3浓度和环境温度之间存在显著的正相关关系(Shen et al,2010;Wang et al,2012),此外研究还发现O3浓度和交通状况之间也存在着联系(Shen et al,2010)。Xue et al(2017)对关中地区城市、城郊和农村站点的VOCs进行了观测和来源解析,探讨了不同VOCs的O3生成潜势。
基于目前对大气化学及光化学反应的认知,国际上已开发了数个大气化学模式,用于模拟大气O3、NOx及其他重要物质的生成过程。Tie et al(2003)在 TUV(Troposheric ultraviolet-visible)辐射传输模式的基础上开发了fast-TUV模式。该模式能够精确地计算对流层太阳辐射传输过程,并且大大提高TUV的计算效率。fast-TUV模式已加入化学传输模式当中,用来计算云和气溶胶对光解速率和大气化学物质的影响(Li et al,2005)。目前,fast-TUV模式已被应用于数个气相化学机制当中,如CBIV、RADM2和SAPRC等,且具有良好的模拟能力。Li et al(2005)在化学传输模式(CTM)中使用fast-TUV模式模拟了美国休斯顿地区黑碳气溶胶对大气光化学过程和O3生成的影响,研究发现,在休斯顿地区污染最重时,黑碳气溶胶的光学效应使边界层内的J[O3(1D)]和J[NO2]值降低10% — 30%,对应的近地面O3浓度减少5% — 20%。对我国关中地区O3污染的模拟研究虽已有开展(Feng et al,2016),但研究仍然较少。
本文应用WRF-CHEM 模式对2015年7月25日至30日关中地区的一次O3重污染事件进行数值模拟,重点研究污染期间西安市和咸阳市O3前体物的主要来源及其对O3浓度的贡献。
1 模式和方法
1.1 WRF-CHEM模式
WRF-CHEM模式是先进的区域动力-化学耦合传输模式,具体细节描述见:https://www2.acom.ucar.edu/wrf-chem。WRF-CHEM包含动力参数化(风、温、边界层、云等)、传输过程(水平传输、垂直输送、扩散等)、干沉降、湿沉降、气相化学、辐射和光化学,并且在线耦合了生物排放。
该报告的模式模拟工作都是基于WRF-CHEM模式(Grell et al,2005)。中国科学院气溶胶化学和物理重点实验室李国辉研究员等对其进一步研发,使其适应中国大气污染的模拟研究,主要发展包括:
(1)发展了一个灵活的气相光化学模式,可以在模拟时使用不同的化学机制(CBIV,RAMD2及SAPRC)。
(2)将美国环保局发展的CMAQ气溶胶模式加入到WRF-CHEM模式。
(3)有机气溶胶的模拟利用非传统的VBS(Volatility basis set)方法,并且包括乙二醛(glyoxal)和甲基乙二醛(methylglyoxal)对二次有机气溶胶的贡献。
(4) 无机气溶胶的模拟利用ISORROPIA Version 1.7。利用西安10年观测的PM2.5组份资料,发展了一个SO2非均相反应的参数化模块,可以更加合理模拟硫酸盐的时空分布。
(5)利用多个城市的观测资料,发展了一个HONO参数化方案,显著改进了模式氧化能力的模拟。
(6)发展了一个计算气溶胶辐射属性(包括光学厚度、单次散射反照率、和奇异性因子)的模块,可以考虑气溶胶对辐射传输的影响。
(7)发展了一个包括气溶胶活化成核的云参数化方案,可以考虑气溶胶对云的影响。
该版本的WRF-CHEM模式已成功模拟了臭氧、无机气溶胶、有机气溶胶、气溶胶的光学厚度和单次散射反照率,气溶胶对云的形成和发展等(Li et al,2008,2009,2010,2011a,2011b,2012)。
模式的模拟结果也会存在一定的不确定性,主要来自以下三个方面:(1)气象场的不确定性,气象条件可以影响污染物的传输以及清除过程,气象场的不确定性是导致污染物模拟偏差的重要原因,引起的模拟偏差能够达到20% — 30%(Bei et al,2017);(2)排放清单的不确定性,排放清单不能及时反映实际排放的快速发展、变化,由此造成的不确定性达10% — 20%(Lei et al,2004;Song et al,2010);(3)模式化学机制也是造成模拟结果不确定性的重要因素(Carter and Atkinson,1996)。
1.2 模式设置
模式模拟所选时段是2015年7月25 — 30日。在所模拟的6天中,7月25、26和29日西安市13个国控站和咸阳市4个国控站平均观测得到的O3最大 1 小时浓度均在 200 µg · m−3以上,污染情况比较严重,7月30日的最大1小时浓度也接近200 µg · m−3,而 7 月 27 和 28 日观测得到的 O3最大 1 小时浓度也高达 150 µg · m−3,但是优于其他4天。模式模拟区域见图1a,中心点是109°E,34.25°N,模拟区域面积为150 km×150 km,水平分辨率6 km。西安市地处关中盆地,关中盆地南倚秦岭,北界陕北高原,西起宝鸡峡,东迄潼关港口,西窄东宽。整个盆地的地形非常不利于污染物的扩散。图1b中的黑色正方形表示关中盆地国控站的位置,其中宝鸡8个站,西安13个站,咸阳4个站,铜川4个站,渭南4个站;图1b中红色圆点代表西安市的中心经纬度。基本上国控站都位于城市中,分布比较密集,本报告主要关注西安和咸阳城市群。
气象场初始和边界条件利用 NCEP FNL 1°×1°资料。化学场初始和边界条件利用 MOZART每6小时的输出。模式模拟采用SAPRC99 化学机制。排放清单由清华大学提供(Zhang et al,2009),其中包括5个部分:农业、工业、能源生产、居民生活及交通运输排放。生物排放源利用在线的MEGAN 模式 Guenther et al(2006)计算。
1.3 统计方法
为更好地评估模式的模拟结果,引入3个统计参数,包括归一化平均偏差(Normalized mean bias,NMB)、 均 方 根 误 差(Root mean square error,RMSE)和 一 致性 指 数(Index of agreement,IOA),定义如下:

其中:Pi和Oi代表模拟和观测变量,N是用于比较的总的时间点数,O代表观测变量的平均值。IOA的变化范围是从0到1,如果IOA等于1,表明模拟和观测非常好地吻合。
1.4 分析方法
大气污染物的形成是一个复杂的非线性过程,受到不同的排放源以及输送过程的影响。模式研究通常用来分析不同因子对污染物形成的贡献,为减排措施提供依据。但是在一个非线性过程中不能直接评估不同因子的影响。某个因子对一个过程的影响,可以分解为该因子的直接贡献和间接贡献(该因子与其它因子相互作用所造成的影响)。因子分析法(Factor separation approach,FSA)常常用来分析不同因子的贡献以及它们之间的相互作用的影响。
例如,假设影响PM2.5形成有两个因子X和Y,并且X和Y相互作用。fXY,fX,fY和 f0分别表示模式模拟包括因子X和Y,只包括X,只包括Y,和不包括因子X及Y。因子X和Y的直接贡献(和)可以表示为:

包括因子X和Y的模拟fXY可以表示为:

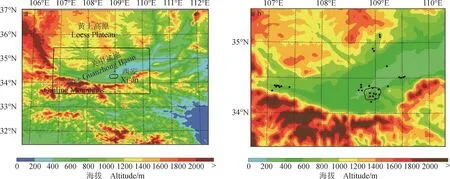
图 1 (a) 模式模拟区域及(b)观测站点分布Fig.1 (a) Simulation domain and (b) observational stations
2 结合和讨论
2.1 模式表现
图2分别给出的是在西安13个国控站和咸阳4个国控站平均的O3和NO2逐日变化模拟与观测比较。从总体上分析,模式能够准确模拟O3浓度的日变化趋势,IOA高达0.98,模式可以准确模拟出下午由于光化学反应产生的O3高值以及夜间与NO反应造成的浓度低值,但是整体上模式略高估O3浓度,6天平均观测得到的O3质量浓度为87.8 µg · m−3,而模拟为 95.1 µg · m−3,这种高估多在夜间产生,可能是由夜间NOx排放的不确定性造成。例如在模式模拟所用的排放清单中,没有包括夜间重型卡车增加所造成的NOx排放。对于NO2的模拟,模式总体上低估NO2的质量浓度,观测到的6天平均NO2的质量浓度为42.0 µg · m−3, 而 模 拟 为 38.0 µg · m−3, 特 别 是 在7月26日上午,模式低估NO2的质量浓度达到30 — 40 µg · m−3。尽管如此,模式能够准确模拟出NO2的日变化趋势,IOA达到0.83。
图3和图4分别给出的是模式模拟及观测的近地面O3在模拟时段内08:00和15:00 LT(Local time,当地时间)的空间分布。在7月25、26、29和30日,模拟的盆地内的水平风速较弱,西安地区有时处于静风状态,不利于污染物的扩散,加之夏季温度较高,有利于O3的产生,模拟的西安的 O3质量浓度在下午 3 时高达 200 µg · m−3,与观测对比模拟结果较好。但是在7月25日和29日存在低估,城市群内某些站点O3的质量浓度变化超过 250 µg · m−3,但是模式没有模拟出这种变化,分析原因可能在于模式的水平分辨率无法分辨出观测到的O3的质量浓度变化;在7月27和28日,由于阴天,云的影响造成O3的质量浓度有所下降,但是在某些地区存在高估的现象,如7月 27 日观测的 O3浓度在 120 µg · m−3左右,而模拟结果在 200 µg · m−3左右,其原因是由于模式不能很好的分辨云系统导致模拟出现高估。在上午8时,由于与氮氧化物的快速反应,O3呈现浓度低值,在7月25、28和30日,模式能够准确模拟出西安市和咸阳市O3的质量浓度变化,但是在7月26、27和29日几乎都存在高估或低估的现象,分析原因可能在于对水平风场模拟的偏差造成的。
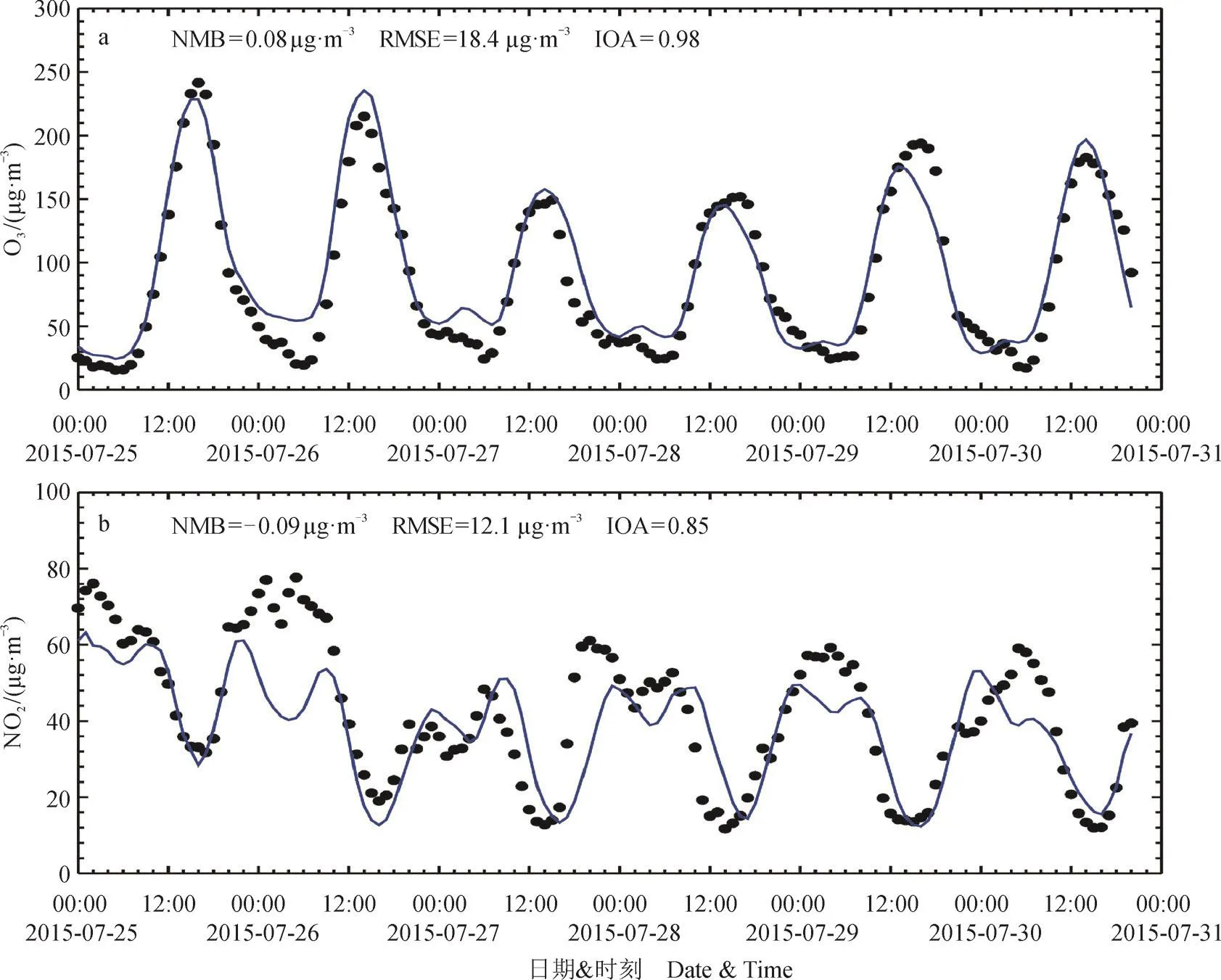
图 2 2015年7月25 — 30日模拟(蓝线)和观测(黑点)的西安市和咸阳市小时平均的 (a) O3和(b) NO2质量浓度Fig.2 Simulated (blue curve) and observed (black spots) (a) O3 and (b) NO2 concentration over Xi'an and Xianyang during the period from 25 to 30 July 2015
图5 和图6分别给出的是模式模拟及观测的近地面NO2在模拟时段内08:00和15:00 LT的空间分布。在上午8时,由于出行高峰,城市内NO2的质量浓度较高,与观测相比,模式倾向于低估。例如在7月26日和30日,西安市内观测得到的NO2的质量浓度超过 50 µg · m−3,而模式没有很好地模拟出NO2质量浓度的这种变化,分析原因可能是模式分辨率的问题,另一种可能的原因是模式没有很准确地模拟出城市尺度边界层高度的变化。在下午3时,此时光化学反应较强,边界层迅速升高,NO2的质量浓度迅速下降,但是与观测相比,仍然存在低估的现象,尤其是在7月25日和26日,观测的 NO2的质量浓度高达 40 µg · m−3,而模式并没有模拟出这种变化趋势。
WRF-CHEM模式基本上可以模拟出西安市和咸阳市O3和NO2的质量浓度的时空分布,但仍然存在偏差。除气象因素以外(如边界层高度和水平风场的模拟),排放源清单的不确定性也是造成模式模拟结果出现偏差的一个重要原因。
2.2 敏感性试验
首先需要关注的问题是各种排放源对西安市和咸阳市城市群O3的贡献,共设计了6个敏感性试验:fall表示所有的排放;fbio表示不包括生物源排放;find表示不包括工业源排放;fpow表示不包括能源生产源排放;fres表示不包括居民源排放;ftra表示不包括交通源排放。若估算交通源对西安市和咸阳市O3的贡献,可通过fall− ftra计算得到。根据研究时段内O3质量浓度的变化情况,将7月25、26、29和30日划分为高浓度臭氧时段,7月27和28日作为低浓度臭氧时段,分别研究臭氧质量浓度不同时段,各个排放源对西安市O3质量浓度的贡献。

图 3 2015年7月25 — 30日在08:00 LT模拟(彩色阴影)和观测(彩色方块)的O3质量浓度Fig.3 Simulated and observed O3 mass concentration at 08:00 LT during the period from 25 to 30 July 2015
图7 是高浓度臭氧时段日平均的各个排放源对西安和咸阳城市群O3的贡献。图7a是日平均生物源对西安市O3的贡献,在臭氧生成的峰值期(12:00 — 18:00 LT),生物源对O3的贡献基本低于25 µg · m−3,平均贡献可达 16.1 µg · m−3(表 1),而在一天中生物源有助于O3的质量浓度的增加,日平均贡献在 8.8 µg · m−3,贡献峰值出现在中午;图 7b是日平均工业源对O3的贡献,可以看出工业源仅在臭氧生成峰值期对臭氧的质量浓度有所贡献,最大贡献量可达到32 µg · m−3,在臭氧生成峰值期的平均贡献为 22.6 µg · m−3,而在其他时段工业源不利于臭氧质量浓度的增加,尤其是在夜间;图7c是平均能源生产对西安市和咸阳市O3的贡献,能源生产基本不利于臭氧质量浓度的增加,仅在14点至23点之间对臭氧的浓度有所贡献,但是低于 5 µg · m−3;图7d是日平均居民源对西安市和咸阳市O3的贡献,从贡献的日变化趋势可以看出,居民源促进臭氧质量浓度的增加,贡献量峰值出现在中午左右,可达到 14 µg · m−3,日平均贡献在 3.4 µg · m−3左右,在臭氧生成峰值期间平均贡献为 7.9 µg · m−3;图 7e 是日平均交通源对西安和咸阳城市群O3的贡献,从8点至24点,伴随着光化学反应的开始,交通源所排放的氮氧化物对O3的质量浓度升高一直有促进作用,在臭氧生成的峰值期,交通源对O3的贡献高达 33 µg · m−3左右,且贡献量一直高于 15 µg · m−3,可见交通源在臭氧生成的峰值期对O3质量浓度的影响很大,平均贡献量为 24.1 µg · m−3,在夜间由于氮氧化物的“滴定作用”,O3有所损耗,交通源不利于O3的生成。由此可以得出,在高臭氧量时段,交通源和工业源在臭氧峰值期的平均贡献最大,且交通源贡献略高于工业源,而且交通源对O3质量浓度的影响可以从清晨一直持续到夜晚。有研究表明(Feng et al,2016),在2013年工业源是西安及周边城市O3的主要来源,而随着近年来交通排放量的增加,交通源对西安市O3质量浓度的影响日益显著,已经超过工业源的贡献。

图 4 2015年7月25 — 30日在15:00 LT模拟(彩色阴影)和观测(彩色方块)的O3质量浓度Fig.4 Simulated and observed O3 mass concentration at 15:00 LT during the period from 25 to 30 July 2015
图8 是低浓度臭氧时段日平均的各个排放源对西安和咸阳城市群O3的贡献。图8a是日平均生物源对O3的贡献,与高臭氧浓度条件下变化趋势相近,在臭氧生成的峰值期(12:00 — 18:00 LT),贡献量变化于 10 µg · m−3和 25 µg · m−3之间,与高浓度臭氧时期相比,日平均贡献量略降低,达到8.2 µg · m−3,但在臭氧峰值期的平均贡献量有所升高,为 17.3 µg · m−3,生物源有助于城市 O3浓度的增加;图8b是日平均工业源对城市O3的贡献,与高浓度臭氧时期相同,工业源在臭氧峰值时期对 O3浓度贡献下降,贡献量低于 20 µg · m−3,平均贡献量为 11.6 µg · m−3;图 8c 是平均能源生产源对西安市和咸阳市O3的贡献,能源生产源几乎不利于臭氧质量浓度的增加,但是与高浓度臭氧时期不同的是能源生产源仅在13点至19点对O3生成有贡献,且贡献量低于 5 µg · m−3;图 8d是日平均居民源对西安和咸阳城市群O3的贡献,从贡献的日变化趋势可以看出,居民源有利于臭氧质量浓度的增加,与高浓度臭氧时期相比,变化不明显,贡献量峰值出现在中午12时,可达到10 µg · m−3左右,日平均贡献在 2.3 µg · m−3左右,在臭氧生成峰值期的平均贡献量为 5.1 µg · m−3;图8e是日平均交通源对西安市和咸阳市城市群O3的贡献,从上午8点开始至23点,交通源开始促进O3的生成,在臭氧生成的峰值期,交 通 源 对 O3的 贡 献 处 于 15 — 30 µg · m−3, 对O3生成的贡献量一直处于高值,平均贡献量为24.7 µg · m−3,与高浓度臭氧时期相比,夜间交通源对O3的降低作用减弱。由此得出,在低臭氧时段,交通源为臭氧生成峰值期最主要的O3贡献源,交通源在臭氧生成峰值期造成的O3质量浓度的增加高出生物源 7 µg · m−3左右,高出工业源约 13 µg · m−3,交通源显著影响城市 O3质量浓度的变化。

图 5 2015年7月25 — 30日在08:00 LT模拟(彩色阴影)和观测(彩色方块)的NO2质量浓度Fig.5 Simulated and observed NO2 mass concentration at 08:00 LT during the period from 25 to 30 July 2015
在臭氧生成的峰值期,由于交通源对O3质量浓度的显著影响,另外需要关注的问题是交通源排放量对西安和咸阳城市群O3质量浓度的影响。随着科技进步和生活水平的提高,人们的生活节奏逐渐加快,交通源已经成为影响空气质量的重要污染源,也是影响城市O3质量浓度的关键因素,在未来的发展中,交通源排放量必然会有所增加。通过上一步的敏感性试验分析,可以知道交通源是臭氧生成峰值期最主要的排放源,为研究交通源对西安和咸阳城市群O3质量浓度的影响,可以设计3个敏感性试验:fall表示基本排放,包括所有排放源;f1.5tra表示在基本排放条件下,将交通源排放量增加为1.5倍;f2.0tra表示在基本排放条件下,将交通源排放量增加为2倍。若估算1.5倍交通源排放量条件下,臭氧的相对增加量可以通过f1.5tra− fall计算得到。

图 6 2015年7月25 — 30日在15:00 LT模拟(彩色阴影)和观测(彩色方块)的NO2质量浓度Fig.6 Simulated and observed NO2 mass concentration at 15:00 LT during the period from 25 to 30 July 2015
图9 给出的是将交通源排放量增加为基本排放的1.5倍和2倍条件下,O3质量浓度的相对增加量。从图中可以看出,随着交通源排放量的增加,在臭氧生成峰值期,西安和咸阳城市群的O3质量浓度也会随之升高。图9a给出的是在整个研究时段内,交通源排放量对O3质量浓度的影响,可以看出交通源排放量的增加在臭氧峰值期间的影响最明显,f2.0tra在峰值期间造成的臭氧增量比f1.5tra高出 10 µg · m−3,当交通源排放量增加 2 倍时,O3质量浓度增量的最大值可以达到 30 µg · m−3,1.5 倍交通源排放量时O3质量浓度增量的最大值也高达20 µg · m−3。表 2 给出的是西安和咸阳城市群,交通源排放量的增加对O3质量浓度的影响。在臭氧生成的峰值期,当交通源排放量增加为1.5倍时,平均的臭氧增量为 14.6 µg · m−3,而当交通源排放量增加为 2 倍时,平均的臭氧增量为 25.4 µg · m−3,可见随着交通源排放量的增加,O3质量浓度迅速增加;图9b和图9c分别给出的是低臭氧时段和高臭氧时段,交通源排放量对O3质量浓度的影响,在臭氧生成的峰值期,f1.5tra造成的平均臭氧增量在低浓度和高浓度臭氧时段分别为10.4 µg · m−3和 16.7 µg · m−3,f2.0tra造 成 的 平 均 臭 氧 增 量 在 低浓 度 和 高 浓 度 臭 氧 时 段 分 别 为 20.7 µg · m−3和27.8 µg · m−3,可见在高浓度臭氧条件下,交通源排放量的增加对O3质量浓度的影响更加显著,在高臭氧时段,f2.0tra造成的臭氧质量浓度的最大增量高于30 µg · m−3,f1.5tra造成的臭氧质量浓度最大增量也高于 20 µg · m−3。城市 O3质量浓度的迅速上升与交通源排放量密切相关,尤其是在O3浓度较高的情况下。

图 7 高臭氧浓度时段(7月25、26、29和30日),各个排放源对西安市和咸阳市O3质量浓度贡献的日变化Fig.7 Diurnal cycle of O3 contribution from each anthropogenic source over Xi'an and Xianyang during high O3 periods (25, 26, 29, and 30 July 2015)
为了更加有效地控制城市臭氧,必须深入了解臭氧的生成特征,臭氧的前体物主要是NOx和VOCs,因此可以通过敏感性试验,控制NOx和VOCs的浓度来检验臭氧生成的敏感性。为探究臭氧生成的敏感性,可以设计4个敏感性试验:Base表示基本排放,包括所有排放源;50%NOx表示在基本排放的条件下,将NOx减少50%;50%VOCs表示在基本排放的条件下,将VOCs减少50%;50%ALL表示在基本排放的条件下,将NOx和VOCs同时减少50%。可以通过50%NOx–Base计算得到在NOx减少50%的条件下臭氧浓度改变量。

表1 高臭氧浓度时段和低臭氧浓度时段,各个排放源在臭氧峰值时期(12:00 — 18:00 LT)对西安市和咸阳市O3的平均贡献量Tab.1 Source contributions to O3 over Xi'an and Xianyang during O3 peaks (12:00 — 18:00 LT) in high and low O3 durations, respectively/(µg · m−3)
图10给出了在臭氧前体物减少的情况下,臭氧浓度以及臭氧改变量的日变化。从图中可以看出,当VOCs减少时,臭氧浓度基本会下降,仅在7月25日出现臭氧质量浓度增加,最大减少量出现在 7 月 25 日,可达 51.4 µg · m−3,研究时段内的臭氧平均减少量为 21.7 µg · m−3(表 3);当NOx减少以及两种前体物的浓度同时减少时,臭氧浓度在臭氧生成的峰值期会下降,在研究时段内臭氧减少量在臭氧生成峰值期的平均值分别为9.9 µg · m−3和 25 µg · m−3,但是在其他时刻臭氧浓度却在增加,这种臭氧浓度改变量的时序分布不能明确反映出西安和咸阳城市群臭氧生成的敏感性特征。

图 8 低臭氧浓度时段(7月27和28日),各个排放源对西安市和咸阳市O3质量浓度贡献的日变化Fig.8 Diurnal cycle of O3 contribution from each anthropogenic source over Xi'an and Xianyang during low O3 periods (27 and 28 July 2015)
图11 给出了在臭氧生成的峰值期,研究时段内,臭氧浓度由于前体物减少而造成的质量浓度平均改变量的空间变化以及H2O2/ HNO3比值的平均值在峰值期间的空间分布。当NOx减少50%时,在臭氧生成峰值期,除城市中心臭氧浓度略增加,而且仅在10 µg · m−3以下,其他地区臭氧质量浓度均在下降(图11a);当VOCs减少50%时,城市群内臭氧质量浓度都在下降,而且城市中心臭氧质量浓度下降最多,可达 50 µg · m−3(图 11b);当NOx和VOCs同时减少50%时,可以看到整个城市区域内,臭氧质量浓度都呈现下降趋势,减少量可达 20 µg · m−3以上,仅在少部分区域,臭氧减少量在 15 — 20 µg · m−3(图 11c)。Sillman(1995)提出了H2O2/ HNO3比值法判定臭氧生成的敏感性。若比值小于0.3,则该地区处于VOCs控制区,若大于0.5则属于NOx控制区,比值在0.3 — 0.5,则该地区属于过渡区。图11d给出了研究时段内,在臭氧峰值期西安和咸阳城市群内H2O2/ HNO3比值的平均变化,可以看到在整个研究区域内,H2O2/ HNO3比值均在0.6以上,基本可以判定西安和咸阳城市群属于NOx控制区,探究臭氧生成的敏感性,可以为控制臭氧提供合理依据。

图 9 在不同臭氧浓度条件下,交通源排放量对西安市和咸阳市O3质量浓度影响的平均日变化Fig.9 Diurnal cycle of O3 contribution from transportation emission over Xi'an and Xianyang at different O3 levels
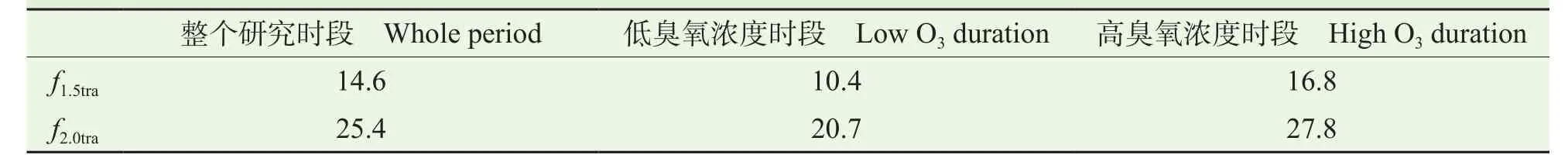
表2 不同臭氧浓度条件下,交通源排放量在臭氧峰值时期(12:00 — 18:00 LT)造成的西安市和咸阳市O3质量浓度平均增加量Tab.2 Peak O3 (12:00 — 18:00 LT) increments over Xi'an and Xianyang owing to transportation emissions at different O3 levels /(µg · m−3)

图10 当西安与咸阳城市群NOx与VOCs排放量分别减少50%以及全部减少50%时,臭氧浓度以及臭氧浓度改变量的日变化Fig.10 Diurnal cycle of O3 changes over Xi'an and Xianyang leaded by a 50% decrease in NOx, VOCs, and both

表3 在臭氧峰值期(12:00 — 18:00 LT),当西安和咸阳城市群NOx与VOCs排放量分别减少50%以及全部减少50%时,臭氧质量浓度的平均改变量Tab.3 Peak O3 (12:00 — 18:00 LT) changes over Xi'an and Xianyang due to a 50% decrease in NOx , VOCs, and both / (µg · m−3)
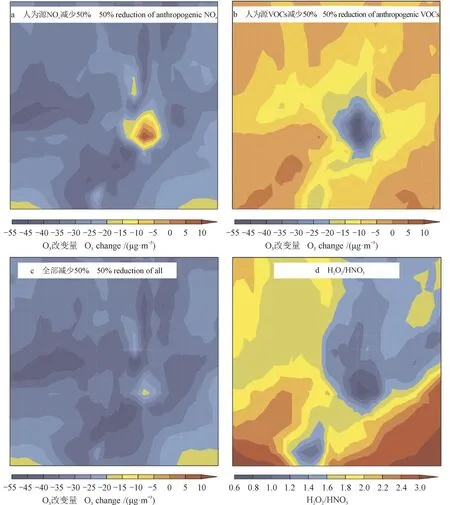
图11 臭氧生成峰值期臭氧浓度改变量以及H2O2 / HNO3的空间变化Fig.11 Spatial distributions of O3 changes leaded by a 50% decrease in (a) NOx , (b) VOCs, and (c) both and (d) H2O2 / HNO3
3 结论
首先需要强调的是,以下所得结论都是基于模式在2015年7月25 — 30日6天的模拟结果,对于不同天气系统下的污染情形不具有代表意义。另外,气象场及排放源的不确定性也会影响模式的评估结果。
(1)WRF-CHEM模式基本上可以合理模拟西安和咸阳城市群O3和NO2的质量浓度的时空分布。但是由于气象场模拟的不确定性及模式分辨率的影响,模拟结果与观测存在偏差。
(2)敏感性试验表明:在臭氧生成的峰值期(12:00 — 18:00 LT),交通源是城市重要的O3源,无论在高浓度臭氧条件下还是低浓度臭氧条件下,贡献量都高于 15 µg · m−3,平均贡献量均高于 24 µg · m−3,而且交通源对 O3的增加作用从早晨8点持续至23点;工业源仅在臭氧峰值生成时期贡献明显,在高臭氧浓度时期,平均贡献量为22.6 µg · m−3,在低臭氧浓度时段,平均贡献量为11.6 µg · m−3,远低于交通源的贡献;从总体上分析,生物源和居民源可以增加O3的质量浓度,在臭氧生成峰值期,生物源无论在高浓度还是低浓度臭氧的条件下,平均贡献都在16 µg · m−3以上,居民源的平均贡献较低,基本低于 10 µg · m−3;能源生产源有降低O3质量浓度的作用,在臭氧生成的峰值时期,能源生产源可以增加O3质量浓度,但是低于 5 µg · m−3。
(3)随着交通源排放量的增加,O3的质量浓度逐渐增加,这种增加主要体现在臭氧的峰值期。当交通源排放量增加至2倍时,O3质量浓度增加量相比于1.5倍的交通源排放量平均高出 10 µg · m−3左右;在臭氧生成的峰值期,当交通源增加为1.5倍时,最大臭氧浓度增量可达到20 µg · m−3, 平均臭氧增量为 14.6 µg · m−3,而当交通源增加为2倍时,最大臭氧浓度增量可达到30 µg · m−3,平均的臭氧增量为 25.4 µg · m−3;在高浓度臭氧条件下,交通源排放量的增加对O3质量浓度的影响更加显著。
(4)在臭氧生成峰值期,当NOx减少50%时,除城市中心臭氧浓度略增加,而且仅在10 µg · m−3以下,其他地区臭氧质量浓度均在下降,平均减少 量 为 9.9 µg · m−3; 当 VOCs 减 少 50% 时, 城市群内臭氧质量浓度都在下降,平均减少量为22 µg · m−3;当 NOx和 VOCs同时减少 50% 时,可以看到整个城市区域内,臭氧质量浓度都呈现下降趋势,减少量可达 20 µg · m−3以上,平均减少量为 23 µg · m−3。在整个研究区域内,H2O2/ HNO3比值均在0.6以上,基本可以判定西安和咸阳城市群属于NOx控制区,探究臭氧生成的敏感性,可以为控制臭氧提供合理依据。
Bei N, Wu J, Feng T, et al. 2017. Impacts of meteorological uncertainties on the haze formation in Beijing-Tianjin-Hebei (BTH) during wintertime: A case study [J].Atmospheric Chemistry and Physics Discussions. DOI:10.5194/acp-2017-698.
Brasseur G P, Orlando J J, Tyndall G S. 1999. Atmospheric chemistry and global change [M]. Cambridge: Oxford University Press.
Cao J, Xu H, Xu Q, et al. 2012. Fine particulate matter constituents and cardiopulmonary mortality in a heavily polluted Chinese city [J]. Environmental Health Perspectives, 120: 373 – 378.
Carter W P L, Atkinson R. 1996. Development and evaluation of a detailed mechanism for the atmospheric reactions of isoprene and NOx[J]. International Journal of Chemical Kinetics, 28:497 – 530.
Cheng H R, Guo H, Saunders S M, et al. 2010. Assessing photochemical ozone formation in the Pearl River Delta with a photochemical trajectory model [J]. Atmospheric Environment, 44: 4199 – 4208.
De Smedt I, Stavrakou T, Müller J F, et al. 2010. Trend detection in satellite observations of formaldehyde tropospheric columns [J]. Geophysical Research Letters,37: L18808. DOI: 10.1029/2010GL044245.
Doherty R M. 2015. Atmospheric chemistry: Ozone pollution from near and far [J]. Nature Geoscience, 8: 664 – 665.
Feng T, Bei N, Huang R J, et al. 2016. Summertime ozone formation in Xi'an and surrounding areas, China [J].Atmospheric Chemistry and Physics, 16: 4323 – 4342.
Geng F, Tie X, Guenther A, et al. 2011. Effect of isoprene emissions from major forests on ozone formation in the city of Shanghai, China [J]. Atmospheric Chemistry and Physics, 11: 10449 – 10459.
Geng F, Zhang Q, Tie X, et al. 2009. Aircraft measurements of O3, NOx, CO, VOCs, and SO2in the Yangtze River Delta region [J]. Atmospheric Environment, 43: 584 – 593.
Grell G A, Peckham S E, Schmitz R, et al. 2005. Fully coupled “online” chemistry within the WRF model [J].Atmospheric Environment, 39: 6957 – 6975.
Guenther A, Karl T, Harley P, et al. 2006. Estimates of global terrestrial isoprene emissions using MEGAN (Model of Emissions of Gases and Aerosols from Nature) [J].Atmospheric Chemistry and Physics, 6: 3181 – 3210.
Lei W, Zhang R, Tie X, et al. 2004. Chemical characterization of ozone formation in the Houston-Galveston area: A chemical transport model study [J]. Journal of Geophysical Research, 109: D12301. DOI: 10.1029/2003JD004219.
Li G, Bei N, Tie X, et al. 2011a. Aerosol effects on the photochemistry in Mexico City during MCMA-2006/MILAGRO campaign [J]. Atmospheric Chemistry and Physics, 11: 5169 – 5182.
Li G, Lei W, Bei N, et al. 2012. Contribution of garbage burning to chloride and PM2.5in Mexico City [J].Atmospheric Chemistry and Physics, 12: 8751 – 8761.
Li G, Lei W, Zavala M, et al. 2010. Impacts of HONO sources on the photochemistry in Mexico City during the MCMA-2006/MILAGO Campaign [J]. Atmospheric Chemistry and Physics, 10: 6551 – 6567.
Li G, Wang Y, Lee K-H, et al. 2009. Impacts of aerosols on the development and precipitation of a mesoscale squall line [J]. Journal of Geophysical Research, 114: D17205.DOI: 10.1029/2008JD011581.
Li G, Wang Y, Zhang R. 2008. Implementation of a twomoment bulk microphysics scheme to the WRF model to investigate aerosol-cloud interaction [J].Journal of Geophysical Research, 113: D15211. DOI:10.1029/2007JD009361.
Li G, Zavala M, Lei W, et al. 2011b. Simulations of organic aerosol concentrations in Mexico City using the WRF-CHEM model during the MCMA-2006/MILAGRO campaign [J].Atmospheric Chemistry and Physics, 11: 3789 – 3809.
Li G, Zhang R, Fan J, et al. 2005. Impacts of black carbon aerosol on photolysis and ozone [J]. Journal of Geophysical Research,110: D23206. DOI: 10.1029/2005JD005898.
Li Y, Lau A K H, Fung J C H, et al. 2013. Importance of NOxcontrol for peak ozone reduction in the Pearl River Delta region [J]. Journal of Geophysical Research: Atmospheres,118: 9428 – 9443.
Lin W, Xu X, Zhang X, et al. 2008. Contributions of pollutants from North China Plain to surface ozone at the Shangdianzi GAW Station [J]. Atmospheric Chemistry and Physics, 8: 5889–5898.
Lu Z, Zhang Q, Streets D G. 2011. Sulfur dioxide and primary carbonaceous aerosol emissions in China and India,1996 — 2010 [J]. Atmospheric Chemistry and Physics, 11:9839 – 9864.
Seinfeld J H, Pandis S N. 2006. Atmospheric chemistry and physics-from air pollution to climate change [M]. 2nd ed.New Jersey: John Wiley & Sons.
Shen Z, Han Y, Cao J, et al. 2010. Characteristics of traff i c-related emissions: a case study in roadside ambient air over Xi'an,China [J]. Aerosol and Air Quality Research, 10: 292 – 300.
Sillman S. 1995. The use of NOy, H2O2, and HNO3as indicators for ozone-NOxhydrocarbon sensitivity in urban locations [J]. Journal of Geophysical Research:Atmospheres, 100: 14175 – 14188.
Song J, Lei W, Bei N, et al. 2010. Ozone response to emission changes: a modeling study during the MCMA-2006/MILAGRO Campaign [J]. Atmospheric Chemistry and Physics, 10: 3827–3846.
Stevenson D S, Dentener F J, Schultz M G, et al. 2006.Multimodel ensemble simulations of present-day and near-future tropospheric ozone [J]. Journal of Geophysical Research, 111: D08301. DOI: 10.1029/2005JD006338.
Tang G, Li X, Wang Y, et al. 2009. Surface ozone trend details and interpretations in Beijing, 2001—2006 [J].Atmospheric Chemistry and Physics, 9: 8813 – 8823.
Tang G, Wang Y, Li X, et al. 2012. Spatial-temporal variations in surface ozone in Northern China as observed during 2009–2010 and possible implications for future air quality control strategies [J]. Atmospheric Chemistry and Physics, 12:2757 – 2776.
Tie X, Geng F, Guenther A, et al. 2013. Megacity impacts on regional ozone formation: observations and WRF-Chem modeling for the MIRAGE-Shanghai fi eld campaign [J].Atmospheric Chemistry and Physics, 13: 5655 – 5669.
Tie X, Geng F, Peng L, et al. 2009. Measurement and modeling of O3variability in Shanghai, China: Application of the WRFChem model [J]. Atmospheric Environment, 43: 4289 – 4302.
Tie X, Madronich S, Walters S, et al. 2003. Effect of clouds on photolysis and oxidants in the troposphere [J].Journal of Geophysical Research, 108: 4642. DOI:10.1029/2003JD003659.
Verstraeten W W, Neu J L, Williams J E, et al. 2015. Rapid increases in tropospheric ozone production and export from China [J]. Nature Geoscience, 8: 690 – 695.
Wang T, Ding A, Gao J, et al. 2006. Strong ozone production in urban plumes from Beijing, China [J]. Geophysical Research Letters, 33: L21806. DOI: 10.1029/2006GL027689.
Wang T, Wei X L, Ding A J, et al. 2009. Increasing surface ozone concentrations in the background atmosphere of Southern China, 1994 — 2007 [J]. Atmospheric Chemistry and Physics, 9: 6217 – 6227.
Wang X, Shen Z, Cao J, et al. 2012. Characteristics of surface ozone at an urban site of Xi'an in Northwest China [J].Journal of Environmental Monitoring, 14: 116 – 126.
Wang X, Zhang Y, Hu Y, et al. 2011. Decoupled direct sensitivity analysis of regional ozone pollution over the Pearl River Delta during the PRIDE-PRD2004 campaign [J].Atmospheric Environment, 45: 4941 – 4949.
Xu J, Ma J Z, Zhang X L, et al. 2011. Measurements of ozone and its precursors in Beijing during summertime: impact of urban plumes on ozone pollution in downwind rural areas [J].Atmospheric Chemistry and Physics, 11: 12241–12252.
Xue L K, Wang T, Gao J, et al. 2014. Ground-level ozone in four Chinese cities: precursors, regional transport and heterogeneous processes [J]. Atmospheric Chemistry and Physics, 14: 13175 – 13188.
Xue Y, Ho S S H, Huang Y, et al. 2017. Source apportionment of VOCs and their impacts on surface ozone in an industry city of Baoji, Northwestern China [J]. Scientif i c Reports, 7.DOI: 10.1038/s41598-017-10631-4.
Zhang Q, Streets D G, Carmichael G R. 2009. Asian emissions in 2006 for the NASA INTEX-B mission [J]. Atmospheric Chemistry and Physics, 9: 5131–5153.
Zhang Y H, Su H, Zhong L J, et al. 2008. Regional ozone pollution and observation-based approach for analyzing ozone-precursor relationship during the PRIDE-PRD2004 campaign [J]. Atmospheric Environment, 42: 6203 – 6218.
Zhou X, Zhang N, TerAvest M, et al. 2011. Nitric acid photolysis on forest canopy surface as a source for tropospheric nitrous acid [J]. Nature Geoscience, 4: 440 – 443.
猜你喜欢
中国环境科学(2023年8期)2023-08-29 09:26:48
作物学报(2022年9期)2022-07-18 00:58:42
湘潮(上半月)(2021年10期)2021-12-02 02:09:38
儿童与健康(幼儿教师参考)(2020年11期)2020-11-25 11:22:18
公民与法治(2020年15期)2020-09-25 02:57:54
中国卫生质量管理(2019年5期)2019-10-24 06:25:24
作文大王·低年级(2018年5期)2018-06-20 05:15:56
知识经济·中国直销(2018年1期)2018-01-31 01:52:37
商周刊(2017年6期)2017-08-22 03:42:37
环境保护与循环经济(2017年9期)2017-03-16 03:15:13
