
基于线粒体多基因串联序列的蚬蝶类系统发生关系分析
2017-12-28 06:54:14石庆会张桂清韦中学
三明学院学报 2017年6期
石庆会,张桂清,韦中学,吴 楠,张 严
(1.三明学院 资源与化工学院,福建 三明365004;2.福建省资源环境监测与可持续经营利用重点实验室,福建 三明 365004;3.福建省矿山生态修复工程技术研究中心,福建 三明 365004)
基于线粒体多基因串联序列的蚬蝶类系统发生关系分析
石庆会1,2,3,张桂清1,韦中学1,吴 楠1,张 严1,2,3
(1.三明学院 资源与化工学院,福建 三明365004;2.福建省资源环境监测与可持续经营利用重点实验室,福建 三明 365004;3.福建省矿山生态修复工程技术研究中心,福建 三明 365004)
测定了蚬蝶类两个代表种(波蚬蝶、银纹尾蚬蝶)的线粒体基因 16S rRNA、Cytb、COI、COIII的部分序列。同时,结合GenBank数据库中已公布的其他蝶类 52个代表种相应的基因序列,选取蛾类 2个代表种作为外类群。基于这4个基因的串联序列,分别使用邻接法、最大简约法、最大似然法和贝叶斯推论法重新构建了蝶类的分子系统树,分析了蚬蝶类的系统发生关系。序列分析结果显示:经比对处理后串联序列共有 4784个同源位点,其中保守位点2113个,变异位点2669个,简约信息位点2149个;A+T平均含量为 76.8%,具有明显的AT偏倚性。分子系统树表明:蚬蝶类形成一个单系群,并与灰蝶科形成姊妹群,结果进一步表明蚬蝶类与灰蝶科具有较近的亲缘关系,但能否归入灰蝶科还有待于进一步的论证。
鳞翅目;蚬蝶科;线粒体基因;系统发生关系
蚬蝶类隶属于鳞翅目、凤蝶总科,是小型或中型美丽的蝴蝶。有的性二型很明显,无季节差异。幼虫宽扁,一般多毛,与灰蝶相似。取食某些药用植物,如千里光及蓟类植物。蚬蝶善于模拟其他蝶类的形态,而且颜色和翅形多种多样,故不易辨认。全世界已知1500多种,中国有 26种,90%以上的种类分布在新热带区,而古北区、东洋区和非洲区种类较少。目前,有关蚬蝶系统发生地位的界定还存在一定的分歧。形态学上,由于蚬蝶类与灰蝶类具有相似的形态特征和生活习性,具有相同的寄主植物,部分学者认为两者具有较近的亲缘关系,并将蚬蝶类视为灰蝶科中的一个亚科[1-3]。然而,其他的形态学研究结果却认为蚬蝶类应作为一个独立的科级分类阶元[4]。近二十多年来,随着 DNA测序与序列分析和分子系统学研究技术的日益成熟,关于蚬蝶类系统发生地位及其与其他蝶类系统发生关系的分子系统学研究也取得了一定的进展。但是,相关研究结果之间还存在较大的争议。主要存在两种观点,一种观点认为蚬蝶应作为一个独立的科级分类阶元,并与灰蝶科形成姊妹群[5-9]。另一种观点却支持将蚬蝶科归入灰蝶科[10-11]。由此可见,关于蚬蝶类的系统发生地位的界定还需要更多分子标记和分析方法的介入,并结合形态学、分子生物学及其他相关学科证据进行综合分析。
线粒体DNA由于具有分子量小、结构简单、母系遗传、几乎不发生重组并且进化速度快等特点而被广泛的应用于动物系统发育与进化、群体遗传学以及比较和功能基因组的研究[12-13]。因此,本文拟通过 PCR扩增技术补充测定蚬蝶类 2个代表种,即波蚬蝶(Zemeros flegyas)(波蚬蝶属)和银纹尾蚬蝶 (Dodona eugenes)(尾蚬蝶属)的线粒体基因 16S rRNA、Cytb、COI、COIII的部分序列。同时,结合已知的其他蝶类主要类群代表种的相应基因序列数据,选择适宜的蛾类代表种作为外类群,运用多种建树方法重建蝶类主要类群的分子系统树,进一步探讨蚬蝶类的系统学地位,从而为蚬蝶类乃至整个蝶类的系统学和分类学研究提供更多新的分子生物学证据。
1 材料与方法
1.1 标本的采集和保存
实验所用的波蚬蝶和银纹尾蚬蝶标本分别采集于江西九连山 (2014年 8月)和四川青城山(2015年5月)。标本采集后参考《中国蝶类志》[14]进行鉴定与登记,再浸泡于无水乙醇中,带回实验室后置于-20℃冰箱中保存备用。
1.2 基因组DNA的提取与纯化
利用动物基因组 DNA快速抽取盒 (生工生物工程 (上海)股份有限公司,型号:B518221-0050)提取基因组 DNA,具体的操作步骤参照试剂盒说明书。
1.3 PCR扩增与测序
实验扩增的4种短基因片段(16S rRNA、Cytb、COI、COIII)均采用通用引物进行扩增。扩增反应体系共 50 μL, 其中包括 Buffer (含 Mg2+) 溶液 8 μL、 BSA 溶液 8 μL、 dNTP 溶液 1.2 μL、DNA1.5~2.5 μL、 正向引物 1 μL、 反向引物 1 μL、Taq酶 0.8 μL 以及灭菌水 ddH2O 若干 (将体系补充至 50 μL)。
扩增反应程序为:95℃预变性 5 min;95℃变性 1 min;退火 (温度根据引物的不同而变化))1 min;72℃延伸 1 min 30 s,循环次数为 35次;最后 72℃总延伸 10 min。 PCR扩增产物利用1.2%琼脂糖凝胶电泳检测,并送至生工生物工程(上海)股份有限公司进行纯化测序。
1.4 数据的收集与整理
本文从 GenBank数据库中共下载了其他蝶类 52个代表种和外类群2个代表种雕尺蛾(Biston panterinaria)、桑弥尺蛾 (Phthonandria atrilineata)的线粒体基因组全序列,并从中截取4个基因(16S rRNA、 Cytb、 COI、 COIII) 的相应序列进行相关分析(表 1)。

表1 分析类群代表种的序列来源信息
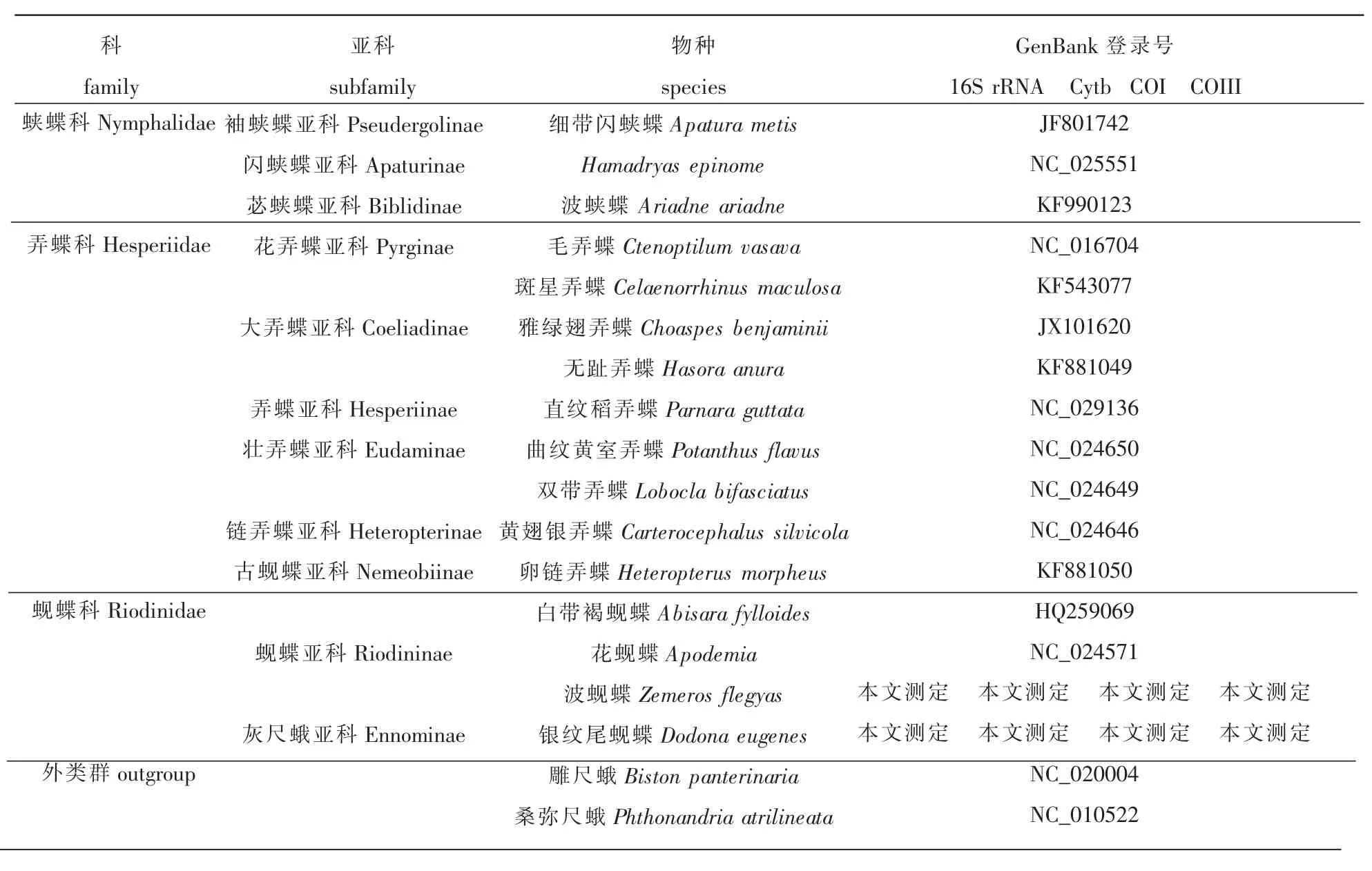
续表1
1.5 数据处理与分析
1.5.1 序列组成与变异分析
将所测定的序列在NCBI网站上运用BLAST功能进行序列比对,确认所测序列为目的基因序列,并确保序列方向一致。运用MEGA 6.0软件[15]进行序列对位排列、序列组成与进化特征分析。
1.5.2 碱基替代饱和度分析
分子数据通常要进行多重替代(multiple substitution)或出现饱和(saturation)现象,受进化噪音影响的可能性较大,从而对系统发育分析产生一定的影响,尤其是距离法或最大简约法分析。因此,对数据进行碱基替代饱和度分析是必要的。本文运用MEGA 6.0[15]软件分别统计数据集中各序列之间的差异度(转换Ts、颠换Tv)和相应的遗传距离(p-distance),并以差异度为纵轴,遗传距离为横轴在Excel中作散点图,判断其是否达到饱和。随着距离的增加,观察到的差异数不再增加时,则认为此序列达到饱和,重建系统发生时要进行特别加权[16]。
1.5.3 遗传距离的计算
遗传距离能够更全面地反映亲本品种间的遗传差异,是衡量品种间若干性状综合遗传差异大小的指标。本文运用MEGA 6.0软件15],将对比过的数据先按照不同类群进行分组,再基于p-distance模型计算组间平均遗传距离。
1.6 分子系统树的构建
分别采用邻接法(neighbor joining,NJ)、最大简约法(maximum parsimony method,MP)、最大似然法(maximum likelihood method,ML)和贝叶斯推论法(Bayesian inference,BI),重新构建蝶类主要类群54个代表种的分子系统树。NJ树通过MEGA 6.0软件[15]构建,具体参数设置如下:Method:p-distance,Variance Estimation Method:Bootstrap method,No.of Bootstrap Replications:1000,Substitutions to Include:d:Transitions+Transversions,系统发生树采用内部分支检验和1000次抽样自展检验来评估各节点的置信度。MP树和ML树分别通过PAUP*4.0b10软件和RaxML在线构建,均采用启发式搜索,系统树各分支的置信度分别以1000次自举检验值和100次自举检验值表示。应用Modeltest 3.7软件在 AIC标准下估算出构建 MP树和ML树时所用的最优模型 (GTR+I+Γ)。BI树利用 MrBayes 3.1.2(Huelsenbeck and Ronquist,2001)构建,所用的模型与构建MP树和ML树时所用到的模型相同,即 lset设置替换模型 nst=6(GTR模型),位点速率变异模型设置为rates=gamma(gamma分布)。同时建立4条马尔科夫链(MCMC chains),以随机树为起始树,共运行25万代,每100代抽样一次,重复1次,直至标准误差小于0.01,每2 500代抽样一次,舍弃老化样本构建一致树。
2 结果
2.1 序列组成与变异
本文分析的54个蝶类代表种的16S rRNA、Cytb、COI和COIII基因序列,经比对剪切后,长度分别为1 372、1 126、1 505和781 bp,4个基因的串联序列总长度为4 784 bp。串联序列中保守位点2 113个,变异位点为2 669个,简约信息位点为2 149个(表2)。
基因序列碱基组成分析结果显示,不论是单个基因还是串联基因序列,A+T平均含量均超过了70%,并明显高于G+C的平均含量,与其他鳞翅目昆虫线粒体基因一样,具有明显的AT偏倚性(表 3)。

表2 4个基因序列及其串联序列的进化特征

表3 4个基因及其串联序列碱基组成
2.2 碱基替换
本研究运用MEGA 6.0软件[15]分别统计了串联序列之间的差异度(转换Ts、颠换Tv)和相应的遗传距离(p-distance),并以差异度为纵轴,遗传距离为横轴在Excel中作散点图如图1所示。结果显示:随着遗传距离的增加,转换与颠换均出现递增的趋势,并保持着良好的线性关系,转换与颠换值均未超过2.0表明碱基替换代未出现饱和状态。因此,串联基因序列转换与颠换的信息都将应用于后续的系统发生树的构建。
2.3 遗传距离分析
采用 MEGA6.0软件[15]基于K2P模型计算6科54种蝶类与外类群之间的遗传距离,结果如表4所示。结果显示:蝶类各科间的遗传距离介于0.118~0.163之间,平均遗传距离为0.134,其中蚬蝶类与灰蝶科的遗传距离最小(0.118),而蚬蝶类与蛱蝶科的遗传距离为0.129。由此可见,本文所选取的蚬蝶类与灰蝶科间的亲缘关系相对于蛱蝶科更为亲近。
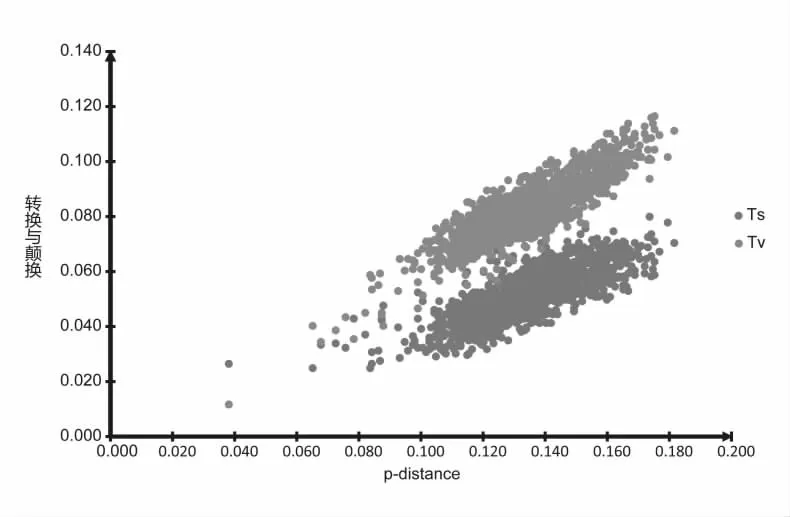
图1 本研究串联基因序列的碱基替代饱和度散点图

表4 基于串联基因序列计算蝶类主要类群间的平均遗传距离
2.4 系统发生关系分析
基于4个基因的串联序列,以蛾类的两个代表种作为外类群,分别应用邻接法、最大简约法、最大似然法和贝叶斯推论法重建了蝶类的分子系统树。结果显示,NJ树、MP树、ML树以及BI树的拓扑结构基本一致,其中ML树和BI树最为稳定,如图3~4所示。
ML树和BI树均显示,蛱蝶科、灰蝶科、蚬蝶科、粉蝶科、弄蝶科、凤蝶科的代表种均形成单系群,其中蚬蝶形成一个独立支系(ML树和BI树的置信值分别为64%和0.94),并与灰蝶科形成稳定的姊妹群(ML树和BI树的置信值分别为100%和1.00)。然而,蛱蝶科并没有直接与灰蝶科形成姊妹群,而是与灰蝶科+蚬蝶科的组合群形成姊妹群 (ML树和BI树的置信值分别为80%和1.00),蝶类主要类群间的系统发生关系可表示为((((蛱蝶科+(灰蝶科+蚬蝶科))+粉蝶科)+弄蝶科)+凤蝶科)。
3 讨论
通过对蝶类6科54个代表种4个线粒体基因(16S rRNA、COI、COIII、Cytb)部分序列及其串联基因序列进行分析,结果显示,4个线粒体基因出现明显的A+T含量偏高的现象,这与其他文献报道的昆虫线粒体基因核苷酸组成频率相一致,均存在富含AT碱基现象[17]。Knight and Mindell(1993)认为转换颠换比的值小于2.0,则此基因序列的突变已达到饱和状态,受进化噪音影响的可能性较大,重建系统发生时不进行特别加权就会得出错误信息[18]。根据已有的研究认为,出现这种结果的原因有两个:第一,可能是由于某些位点发生了多次转换替代,即转换出现了饱和现象;第二,与高A+T含量有很大的关系,由于高A+T含量增加了颠换的可能性[19]。本文序列分析结果显示,无论是单个基因序列还是串联序列,均出现富含A+T碱基的现象,究其原因,本文认为是由于高A+T含量导致颠换增加的可能性。本文分子系统树分析结果显示,蚬蝶类的代表种形成单系群,并与灰蝶科具有较近的亲缘关系。同时,组间遗传距离分析结果显示,蚬蝶类与灰蝶科的遗传距离最小,同样表明两者之间具有较近的亲缘关系,这与前期基于分子证据的部分研究结果相吻合[5-9]。然而,本文所选取的蚬蝶类代表种存在一定的局限性,未能涵盖蚬蝶所有的分类群,因此并不能直接认定蚬蝶类可以作为灰蝶科内的一个亚科级分类阶元,并不支持前期基于形态学证据[1-3]和部分分子证据[10-11]的研究结果。

图2 基于串联基因序列构建的蝶类主要类群的ML树
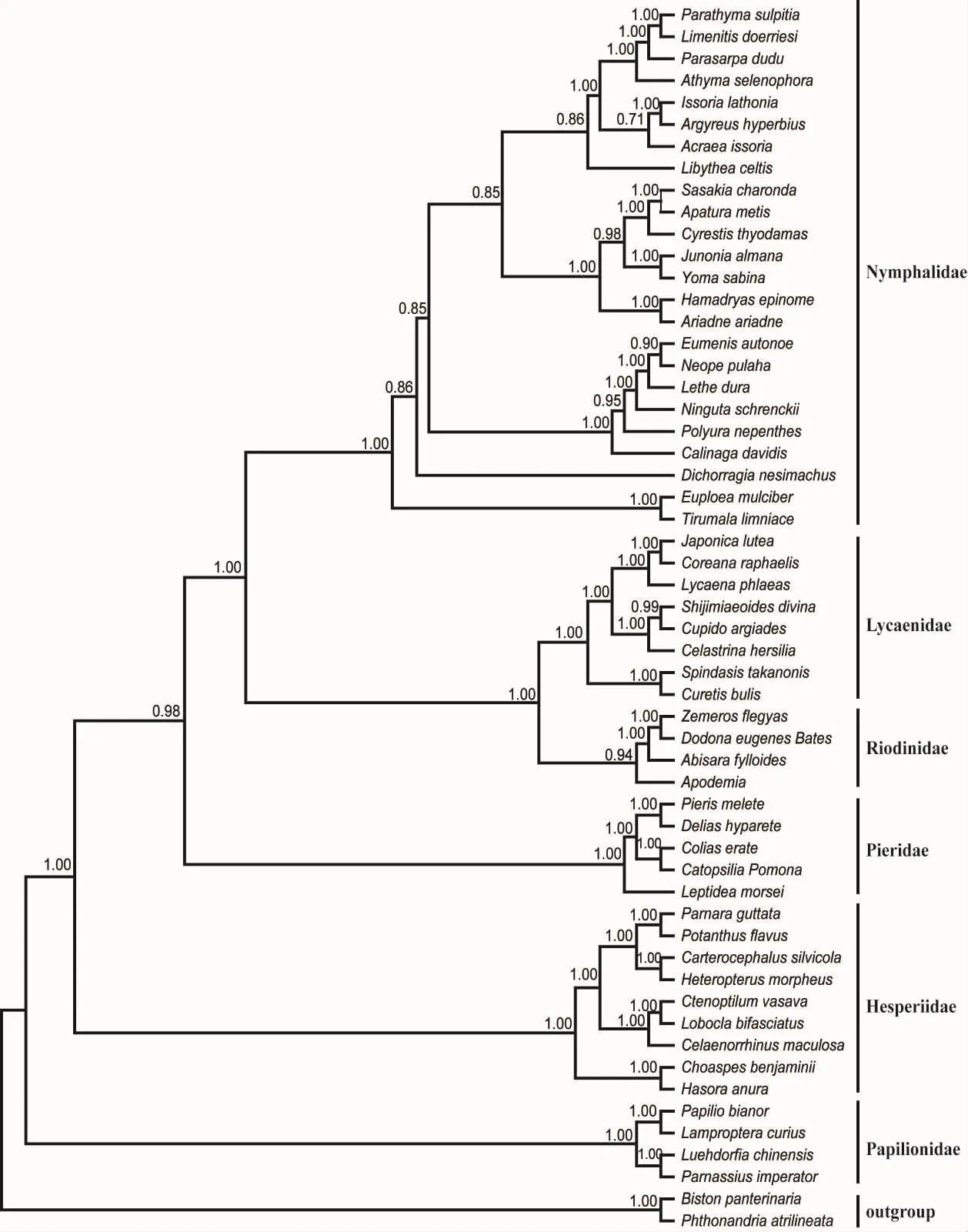
图3 基于串联基因序列构建的蝶类主要类群的BI树
综上所述,本文认为蚬蝶类还是作为一个科级分类阶元--蚬蝶科较为合理,并与灰蝶科具有较近的亲缘关系。至于能否将其作为一个亚科级分类阶元归入灰蝶科,还有待于更多蚬蝶代表种以及更多分子证据的的介入进行更充分的论证。
[1]KRISTENSEN N P.Remarks on the family-level phylogeny of butterflies (Insecta, Lepidoptera, Rhopalocera)[J].J Zool Syst Evol Res,1976,14:25-33.
[2]ACKERY PR.Systematic and faunistic studies on butterflies [M]//Vane-Wright R I,Ackery PR.The Biology of Butterflies London:Academic Press,1984.
[3]周尧.中国蝴蝶分类与鉴定[M].郑州:河南科学技术出版社,1998.
[4]寿建新,周尧,李宇飞.世界蝴蝶分类名录[M].西安:陕西科技出版社,2006.
[5]WELLER S J,PASHLEY D P,MARTIN J A.Reassessment of butterfly family relationships using independent genes and morphology[J].Ann Entomol Soc Am,1996,89:184-192.
[6]CAMPBELL D L,BROWER A V Z, PIERCE N E.Molecular evolution of the wingless gene and its implications for the phylogenetic placement of the butterfly family Riodinidae (Lepidoptera:Papilionoidea)[J].Mol Biol Evol,2000,17:684-696.
[7]WAHLBERG N,BRABY M F,BROWER A V Z,et al.Synergistic effects of combining morphological and molecular data in resolving the phylogeny of butterflies and skippers[J].Proc R Soc B,2005,272:1577-1586.
[8]HEIKKILa M, KAILA L,MUTANEN M,et al.Cretaceous origin and repeated tertiary diversification of the redefined butterflies[J].Proc R Soc B, 2012,279:1093-1099.
[9]杨邦和,吴孝兵,诸立新,等.基于COII和EF-1ɑ基因部分序列的中国蝶类科间系统发生关系[J].动物学报,2008,54:233-244.
[10]邹方振,郝家胜,黄敦元,等.基于线粒体ND1和16S rRNA基因序列探讨国产12科蝶类的系统发生关系[J].昆虫学报,2009,53:118-126.
[11]ZHAO F,HUANG D Y,SUN X Y,et al.The first mitochondrial genome for the butterfly family Riodinidae (Abisara fylloides) and its systematic implications [J].Zool Res,2013,34:E109-E119.
[12]VIGILAN L,STONEKING M, HARPENDING H.African populations and the evolution of human mitochondrial[J].Science,1991,253:1503-1507.
[13]STONEKING M,SOODYALL H.Human evolution and the mitochondrial genome[J].Curr Opin Genet Dev,1996(6):731-736.
[14]周尧.中国蝶类志[M].郑州:河南科学技术出版社,1999.
[15]TAMURE K,STECHER G,PETER D,et al.MEGA6:Molecular evolutionary genetics analysis sersion 6.0[J].Mol Biol Evol, 2013, 30:2725-2729
[16]寿建新.国内外蝴蝶分类认识总结[J].西安文理学院学报(自然科学版),2014(4):12-13.
[17]夏靖,胡静,朱国萍,等.大卫绢蛱蝶线粒体基因组全序列的测定和分析[J].昆虫学报,2011(5):34-36.
[18]毛增辉,郝家胜,朱国萍,等.菜粉蝶线粒体基因组全序列的测定和分析[J].昆虫学报,2010,48:2-4.
[19]王菊平,聂新平,曹天文,等.大紫蛱蝶粒体基因组全序列的测定和分析[J].动物分类学报,2012(1):27-28.
Phylogenetic Placement of Riodinids(Lepidoptera:Papilionidae)Based on Multiple Mitochondrial Gene Sequences
SHI Qing-hui1,2,3,ZHANG Gui-qing1,WEI Zhong-xue1,WU Nan1,ZHANG Yan1,2,3
(1.School of Resources and Chemical Engineering,Sanming University,Sanming 365004,China;2.Fujian Provincial Key Laboratory of Resources and Environment Monitoring and Sustainable Management and Utilization,Sanming University,Sanming 365004,China;3.Fujian Province Engineering Research Center of Mine Ecological Construction,Sanming University,Sanming 365004,China)
The partial mitochondrial 16S rRNA,Cytb,COI and COIII genes ofZemeros flegyasandDodona eugeneswere newly amplified and sequenced.Meanwhile,the homologous sequences of 52 representative species of butterfly were obtained from the GenBank.Based on these data,the sequence variation and the phylogenetic relationship of these groups were analyzed,and the phylogenetic trees of the 54 species from all currently recognized families of butterfly were reconstructed based the combined dataset of the four gene sequences with the neighbor joining (NJ),maximum parsimony(MP),maximum likelihood (ML)and Bayesian Inference(BI)methods.The results of the sequence analysis showed that the four combined genes are 4784 bp in length by alignment,including 2113 conserved sites,2669 variable sites and 2149 parsimonious-informative sites,and the average percentage of A+T is 76.8%.The results of phylogenetic analyses indicated that the riodinid butterflies were strongly supported monophyletic groups and sister to Lycaenidae.Therefore,the results of this paper further confirmed that the Riodinidae and Lycaenidae are more closely to each other.However,it is needed to further prove whether it belongs to Lycaenidae.
lepidoptera;riodinidae;mitochondrial gene;phylogeny
Q969.432.1
A
1673-4343(2017)06-0009-09
10.14098/j.cn35-1288/z.2017.06.002
2017-09-29
福建省教育厅中青年教师教育科研项目(JAT160456);三明学院科学研究基金(B201605);福建省科技厅自然科学基金项目(2017J05059);福建省自然科学基金项目(2014J01136)
石庆会,男,安徽宿松人,讲师,博士。主要研究方向:昆虫资源监测、分子系统学与进化生态学。
朱联九)
猜你喜欢
故事作文·低年级(2023年11期)2023-12-05 06:39:56
故事作文·低年级(2023年12期)2023-03-24 14:16:52
园艺与种苗(2021年10期)2021-11-25 05:05:22
生物灾害科学(2021年1期)2021-04-09 07:04:12
科学导报(2020年62期)2020-09-29 09:16:52
江苏农业科学(2019年5期)2019-09-02 14:01:46
广东农业科学(2017年5期)2017-08-29 10:37:31
中国环境监察(2016年7期)2016-10-23 05:36:30
中国现当代社会文化访谈录(2016年0期)2016-09-26 08:46:23
应用海洋学学报(2014年1期)2014-11-22 07:17:44
