
QuEChERS-四极杆/静电场轨道阱高分辨质谱测定动物性食品中氟虫腈及其代谢物残留
2017-12-27 02:46:06尹帅星陈达炜方从容赵云峰
分析测试学报 2017年12期
吕 冰,尹帅星,陈达炜*,方从容,周 爽,赵云峰
(1.国家食品安全风险评估中心/卫生部食品安全风险评估重点实验室,北京 100021;2.岛津技迩(上海)商贸有限公司,北京 100027)
QuEChERS-四极杆/静电场轨道阱高分辨质谱测定动物性食品中氟虫腈及其代谢物残留
吕 冰1,尹帅星2,陈达炜1*,方从容1,周 爽1,赵云峰1
(1.国家食品安全风险评估中心/卫生部食品安全风险评估重点实验室,北京 100021;2.岛津技迩(上海)商贸有限公司,北京 100027)
基于低温处理和QuEChERS方法,采用超高效液相色谱-四极杆/静电场轨道阱高分辨质谱建立了动物性食品中氟虫腈及其代谢物残留的分析检测方法。样品采用乙腈提取,经低温处理,N-丙基乙二胺(PSA)和C18粉分散固相萃取(d-SPE)净化,以BEH C18色谱柱为分析柱,乙腈-0.1%乙酸溶液为流动相进行梯度洗脱分离,外标法定量。采用高分辨质谱平行反应监测(PRM)扫描模式,以负离子采集进行定性筛查和定量分析。氟虫腈及其代谢物在0.02~2 μg/L和0.2~20 μg/L质量浓度范围内均呈良好的线性关系,相关系数(r2)大于0.992。对液体或半液体样品(如牛奶和鸡蛋)和固体样品(如鸡肉),方法的定量下限分别为0.1 μg/kg和 0.2 μg/kg。在不同浓度的加标水平下,氟虫腈及其代谢物在鸡蛋中的平均回收率为81.6%~96.9%,相对标准偏差(RSD)为1.3%~11.5%;在鸡肉中的平均回收率为81.2%~96.0%,RSD为3.4%~11.4%;在牛奶中的平均回收率为79.1%~100.1%,RSD为1.5%~10.7%。该方法简单、灵敏、准确,适用于动物性食品中氟虫腈及其代谢物的快速筛查和定量检测,方法的灵敏度满足欧盟的残留限量要求。
氟虫腈;动物性食品;QuEChERS;高分辨质谱;代谢物
氟虫腈是一种苯基吡唑类杀虫剂,主要用于杀灭鳞翅目和直翅目的害虫以及在土壤中的鞘翅目害虫的幼虫,同时用以消除蚤、虱、蜱、蟑螂及螨等昆虫[1]。近期,欧洲多国相继报道在鸡蛋中超标检出杀虫剂“氟虫腈”事件,并导致欧盟多国近千万枚鸡蛋下架[2]。由于短期大量摄入氟虫腈会对神经系统产生不良影响,而长期摄取氟虫腈或会导致肝脏、甲状腺等受损[3-4],欧盟已明令禁止氟虫腈用于人类食品产业链的畜禽养殖过程,并规定氟虫腈(氟虫腈和氟虫腈砜之和,以氟虫腈计)在鸡蛋中的最大残留限量(MRL)为0.005 mg/kg。国际食品法典委员会(CAC)规定氟虫腈在肉、蛋、奶中的MRL均为0.02 mg/kg。我国也制定了氟虫腈在蔬菜、谷类等制品中的MRL,并将氟虫腈的残留物定义为氟虫腈、氟甲腈(MB46513)、氟虫腈砜(MB46136)、氟虫腈亚砜(MB45950)之和[5],但暂无在动物性食品中的MRL规定。因此,建立动物性食品中氟虫腈及其代谢物的高效、快速的残留检测方法,以应对可能因“氟虫腈”事件引发的鸡蛋、鸡肉和牛奶等动物性食品中氟虫腈及其代谢物的检测需要,对保障动物性相关食品安全具有重要意义。
氟虫腈及其代谢物的测定方法主要有气相色谱法(GC)[6]、气相色谱-质谱法(GC-MS)[6-8]、液相色谱法(HPLC)[9]及液相色谱-串联质谱法(LC-MS/MS)[10-12]。但这些检测方法均是应用于植物性样品等基质,而动物性样品的检测分析方法鲜见报道。Zhang等[13]利用C18固相萃取柱结合液相色谱-串联质谱技术建立了鸡蛋和鸡肉中氟虫腈的分析方法,但该方法未对氟虫腈的代谢物进行检测。世界粮农组织(FAO)在氟虫腈的评估报告中指出氟虫腈在动物体内的氟虫腈母体绝大部分转化为氟虫腈砜,后者在蛋黄、皮和脂肪中浓度最高[14]。因此,氟虫腈母体在动物性样品中检出含量较低,而氟虫腈砜或其它代谢物将是检测关注的重点。近年来,QTOF及Orbitrap等高分辨质谱检测技术在农兽药等化学污染物的检测分析中得到广泛应用[15-17]。然而,高分辨质谱技术对目标物的定量检测分析主要采用全扫描模式(Full scan,FS),与三重四极杆的多反应监测模式(MRM)相比,无法获得较佳的仪器方法灵敏度。在前期研究中,本课题组应用四极杆/静电场轨道阱高分辨质谱(Q Exactive)的其它定量扫描模式,如靶向单一离子监测(Target single ion monitoring,TSIM)[18-19]和平行反应监测(Parallel reaction monitoring,PRM)[20],改善了仪器方法的灵敏度,可与三重四极杆的MRM模式相媲美。
本研究采用QuEChERS(Quick、Easy、Cheap、Effective、Rugged、Safe)方法,以乙腈作为提取溶剂,经盐析后,提取液经低温冷冻处理和分散固相萃取(d-SPE)净化,基于超高效液相色谱-四极杆/静电场轨道阱高分辨质谱的PRM模式,首次建立了动物性食品中氟虫腈及其代谢物残留的分析方法。方法的灵敏度满足欧盟的残留限量要求,为动物性食品中氟虫腈及其代谢物的高效、快速测定提供了技术保障。
1 实验部分
1.1 仪器、试剂与材料
超高效液相色谱-四极杆-静电场轨道阱高分辨质谱仪(Q-Exactive,美国赛默飞公司)、组织匀浆机(德国博朗公司)、涡旋混合器(美国Scientific Industries公司)、超声波清洗器(宁波科生仪器厂)和冷冻离心机(德国Sigma公司)。
乙腈为色谱纯(美国Fisher Scientific公司),乙酸为HPLC级(美国Tedia公司);氯化钠和无水硫酸钠为分析纯(国药试剂有限公司);d-SPE净化管,内含N-丙基乙二胺(PSA)50 mg、C18粉50 mg和无水硫酸钠250 mg(岛津技迩(上海)商贸有限公司);氟虫腈、氟甲腈、氟虫腈砜和氟虫腈亚砜标准品(纯度>98%)购自德国Dr.Ehrenstorfer公司;实验用水为重蒸水。实际样品(鸡蛋、鸡肉、猪肉、猪肝和牛奶等)采购于北京各大超市和农贸市场。
1.2 标准溶液的配制
分别准确称取氟虫腈及其代谢物标准品0.01 g(精确至0.000 1 g)于不同的10 mL容量瓶中,用乙腈溶解并定容,配成1 000 mg/L的标准储备溶液,于-20 ℃储存。氟虫腈及其代谢物标准混合中间液(10 mg/L)和标准混合使用液(200 μg/L和20 μg/L)由乙腈逐级稀释氟虫腈及其代谢物标准储备液制得。准确吸取氟虫腈及其代谢物标准混合使用液(20 μg/L和200 μg/L)0.01、0.02、0.05、0.1、0.2、0.5、1.0 mL于10 mL容量瓶中,用50%乙腈水溶液定容,即得0.02~2 μg/L和0.2~20 μg/L质量浓度范围的两套标准工作液,临用现配。
1.3 试样制备
取代表性动物性样品,用组织匀浆机充分搅碎打匀,取其中200 g分装入洁净容器中,密封,于-20 ℃保存。
1.4 试样提取
1.4.1液体与半液体试样准确称取样品5.0 g(精确至0.001 g),置于50 mL离心管中,涡旋混合30 s,加入10 mL乙腈,超声提取15 min,加入2 g氯化钠和6 g无水硫酸钠,涡旋混合30 s,以9 500 r/min于4 ℃离心10 min,转移上清液于15 mL离心管,待净化。
1.4.2固体试样准确称取样品2.0 g(精确至0.001 g),置于50 mL离心管中,加水3 mL,涡旋混合30 s,以下操作程序同“1.4.1”,并将上清提取液置于-20 ℃超低温冰箱中低温冷冻处理2 h后,待净化。
1.5 试样净化
准确吸取提取液1 mL于2 mL的 d-SPE净化管中,涡旋混合30 s,取上清液0.5 mL,加水定容至1 mL,过有机微孔滤膜后,待测定。
1.6 仪器分析条件
色谱条件:BEH C18色谱柱(1.7 μm,2.1 mm×100 mm),柱温:40 ℃;流动相:A为0.1%乙酸溶液,B为乙腈,梯度洗脱,流速0.3 mL/min。梯度洗脱程序:0~6 min,55%~70% B;6~7 min,70%~100% B;7~8 min, 100% B;8~8.5 min,100%~55% B;8.5~11 min,55% B。进样体积:5 μL。
质谱参数:采用HESI离子化方式;喷雾电压为3.2 kV;毛细管温度为320 ℃;加热温度为300 ℃;鞘气为40 arb,辅助气为10 arb;扫描模式为平行反应监测(PRM),负离子采集模式;分辨率采用70 000 FWHM,碰撞能量(NCE)为20%。氟虫腈及其代谢物的定性、定量离子对见表1。
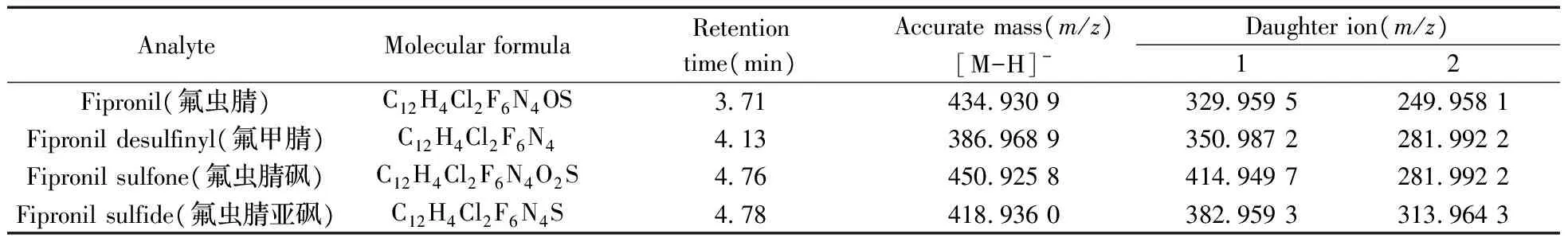
表1 氟虫腈及其代谢物的母离子和子离子信息Table 1 The information for parent and daughter ions of fipronil and its metabolites
2 结果与讨论
2.1 质谱与色谱条件的优化
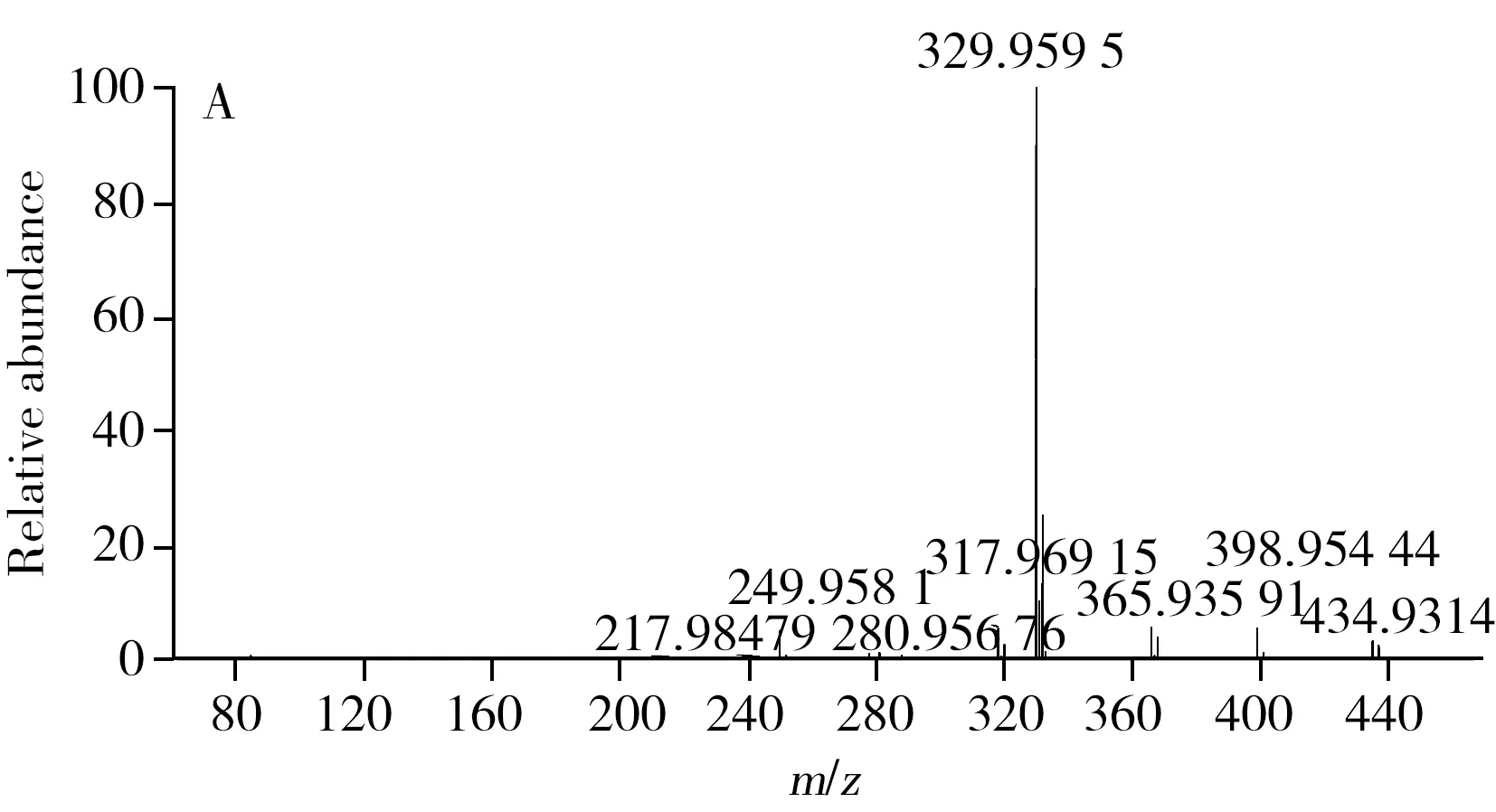
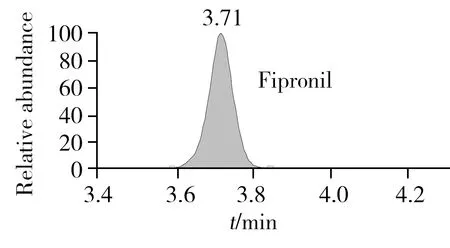
在质谱响应上,氟虫腈及其代谢物的负离子采集模式优于正离子采集模式。本研究比较了Q Exactive高分辨质谱的FS、TSIM和PRM 3种定量采集模式。其中,FS采集模式是高分辨质谱最为常用的定量模式,该模式是对一定质量范围内的所有母离子进行全扫描,TSIM采集模式是对单一母离子进行扫描,而PRM采集模式是先从四极杆中选择目标母离子,然后将其送入高能碰撞池进行二级质谱碎裂,最后以母离子->子离子的精确质量数离子对进行定量,与三重四极杆以离子对为定量手段相似,但其高分辨质谱的检测能力增强了定性确证的能力,同时由于该模式可通过监测其子离子的精确质量数进一步降低背景噪音响应,从而提高了仪器的灵敏度。因此,本研究采用PRM模式对氟虫腈及其代谢物进行定性和定量采集。在PRM模式下,本研究进一步对二级质谱的碰撞能量(NCE)进行优化,结果显示,氟虫腈及其代谢物在20%的NCE下能够获得最佳的二级质谱定量离子响应。图1A为氟虫腈母离子在20%的NCE下所采集的二级质谱图谱,该图谱包含了未被碎裂的母离子精确质量数(m/z434.931 4)与二级质谱子离子的精确质量数(m/z329.959 5,249.958 1),而图1B则为该农药的二级质谱定量子离子(m/z329.959 5)的提取离子色谱图。
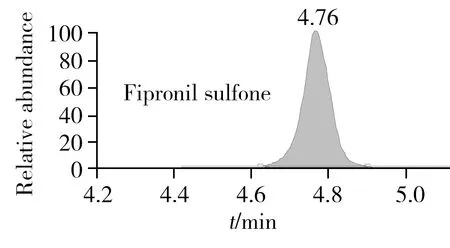
本研究采用BEH C18色谱柱,考察了氟虫腈及其代谢物在乙腈-水、乙腈-0.1%甲酸或乙酸水溶液、乙腈-5 mmol/L甲酸铵或乙酸铵水溶液为流动相体系下的峰形及质谱响应效果。结果表明,氟虫腈及其代谢物在中性或偏碱性流动相体系下峰宽更宽,且氟虫腈砜在酸性流动相体系下具有更好的质谱响应,该现象与文献报道相符[21];而在酸性流动相体系下,鉴于流动相过低的pH值会抑制氟虫腈及其代谢物的响应,因此以乙腈和0.1%乙酸水溶液作为流动相体系。图2为此流动相体系下所获得的氟虫腈及其代谢物的色谱图。
2.2 样品前处理方法的选择
目前,乙腈作为提取溶剂具有较高的沉淀蛋白能力,对亲脂性目标物具有较好的提取效果,同时有助于下一步QuEChERS的净化,因而在动物性食品中具有较好的应用[22],对于固体样品(如鸡肉),需预先加入3 mL 水并对其充分混匀,再用乙腈进行提取,以提高对目标物的提取效率。QuEChERS前处理方法由Anastassiades等[23]于2003年开发,其基于d-SPE技术被广泛应用于农产品中农药多残留检测。本研究采用PSA和C18对样品提取液进行净化,其中PSA能有效去除提取液中的极性物质、有机酸和脂肪酸等杂质;而C18对亲脂性杂质,尤其是脂肪等物质具有较好的吸附效果。此外,本研究取1 mL样品提取液进行净化,50 mg的PSA和50 mg的C18足以达到净化效果,当进一步增加吸附剂填料对净化效果并无明显改善,同时当C18的填料加到100 mg时,对亲脂性的氟虫腈及其代谢物会有一定程度的吸附,导致回收率下降7%~12%。因此,本研究采用内含50 mg的PSA、50 mg的C18和250 mg无水硫酸钠的d-SPE净化管进行净化。
鸡肉等固体动物性食品基质较为复杂,脂肪含量较高,单纯依靠d-SPE净化无法满足去除脂肪的需要。因此为更好地去除脂肪,本研究在进行d-SPE净化前,先将提取液置-20 ℃超低温冰箱中低温冷冻处理,并对冷冻处理时间进行了考察(30 min、1 h、2 h、6 h)。结果发现,提取液冷冻处理2 h后可以达到较好的去除效果,而增加处理时间对去除效果并无明显改进,故选择在d-SPE净化前先对鸡肉提取液进行2 h的冷冻处理。
2.3 基质效应
按照“1.4”和“1.5”方法将试样提取净化,制备鸡蛋、鸡肉和牛奶3种空白基质提取液,对空白基质提取液进行分析,确定其不含有痕量目标化合物。用50%的乙腈水溶液及鸡蛋、鸡肉和牛奶3种空白基质提取液分别配制浓度范围为0.02~2 μg/L和0.2~20 μg/L的两套混合标准溶液并绘制标准曲线。本研究依据基质标准溶液和溶剂标准溶液之间的标准曲线斜率比值评价基质效应,斜率值越接近1.0说明基质效应越弱。当斜率比值在0.8~1.2范围,认为该方法的基质效应可以接受[24]。本方法在低浓度和高浓度标准曲线浓度范围内对氟虫腈及其代谢物的基质效应评价如表2所示,鸡蛋、鸡肉和牛奶的斜率比值范围在0.9~1.1之间,表明本方法中氟虫腈及其代谢物在鸡蛋、鸡肉和牛奶中存在弱的基质效应,基质效应对测定结果的影响可以忽略不计,因而本实验采用溶剂标准曲线进行定量。
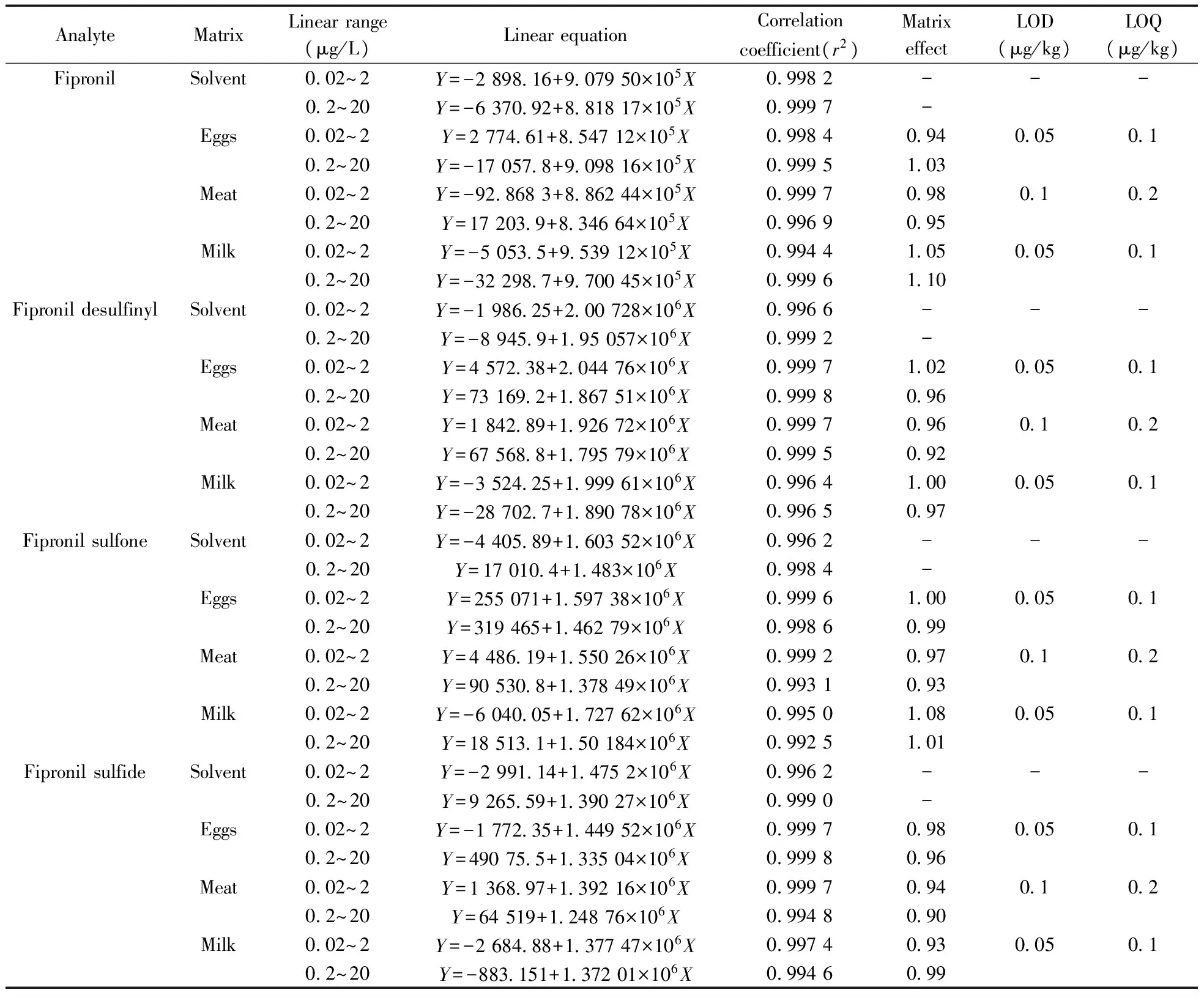
表2 氟虫腈及其代谢物的线性、基质效应、检出限及定量下限Table 2 Linear relationship,matrix effect,LOD and LOQ of fipronil and its metabolites
-:no data
2.4 线性范围与定量下限
本研究配制两套氟虫腈及其代谢物的低高浓度系列混合标准溶液,质量浓度范围分别为0.02~2 μg/L和0.2~20 μg/L,以满足不同浓度水平下实际样品和加标样品的检测。按“1.6”条件进样标准溶液,以标准溶液浓度(X,μg/L)为横坐标,母离子->子离子对的精确质量数的提取离子色谱峰峰面积(Y)为纵坐标,绘制标准曲线,外标法定量。结果表明,氟虫腈及其代谢物在0.02~2 μg/L和0.2~20 μg/L质量浓度范围内均呈良好的线性关系,相关系数(r2)>0.992。以空白样品低水平加标测试方法的检出限和定量下限,以3倍信噪比(S/N=3)对应的浓度为方法检出限,以10倍信噪比(S/N=10)对应的浓度为方法的定量下限。表2结果显示,液体或半液体样品(如牛奶和鸡蛋)取样量5 g时,氟虫腈及其代谢物的方法检出限(LOD)为0.05 μg/kg,定量下限(LOQ)为0.1 μg/kg;固体样品(如鸡肉)取样量2 g时,氟虫腈及其代谢物的方法检出限为0.1 μg/kg,定量下限为0.2 μg/kg。该方法的定量下限均低于欧盟所制定的蛋类中氟虫腈的最大残留限量(5 μg/kg)和CAC所制定的蛋类中氟虫腈的最大残留限量(20 μg/kg)。
2.5 准确度与精密度
本研究取3种代表性样品(鸡蛋、鸡肉和牛奶)进行加标回收实验,以回收率考察方法的准确度,回收率平行测试结果的相对标准偏差(RSD)考察方法的精密度。加标水平的选择以接近定量下限水平的0.2 μg/kg为低加标水平,以欧盟制定的蛋类中氟虫腈的最大残留限量5 μg/kg为中加标水平,以CAC制定的蛋类中氟虫腈的最大残留限量20 μg/kg为高加标水平。取鸡蛋、鸡肉和牛奶3种空白样品分别添加低、中和高加标水平的混合标准溶液后,摇匀,放置一定时间后,按照本方法处理,每个浓度水平重复5次,其回收率和RSD结果见表3。由表3可见,氟虫腈及其代谢物在鸡蛋中的平均回收率为81.6%~96.9%,RSD为1.3%~11.5%;在鸡肉中的平均回收率为81.2%~96.0%,RSD为3.4%~11.4%;而在牛奶中的平均回收率为79.1%~100.1%,RSD为1.5%~10.7%。
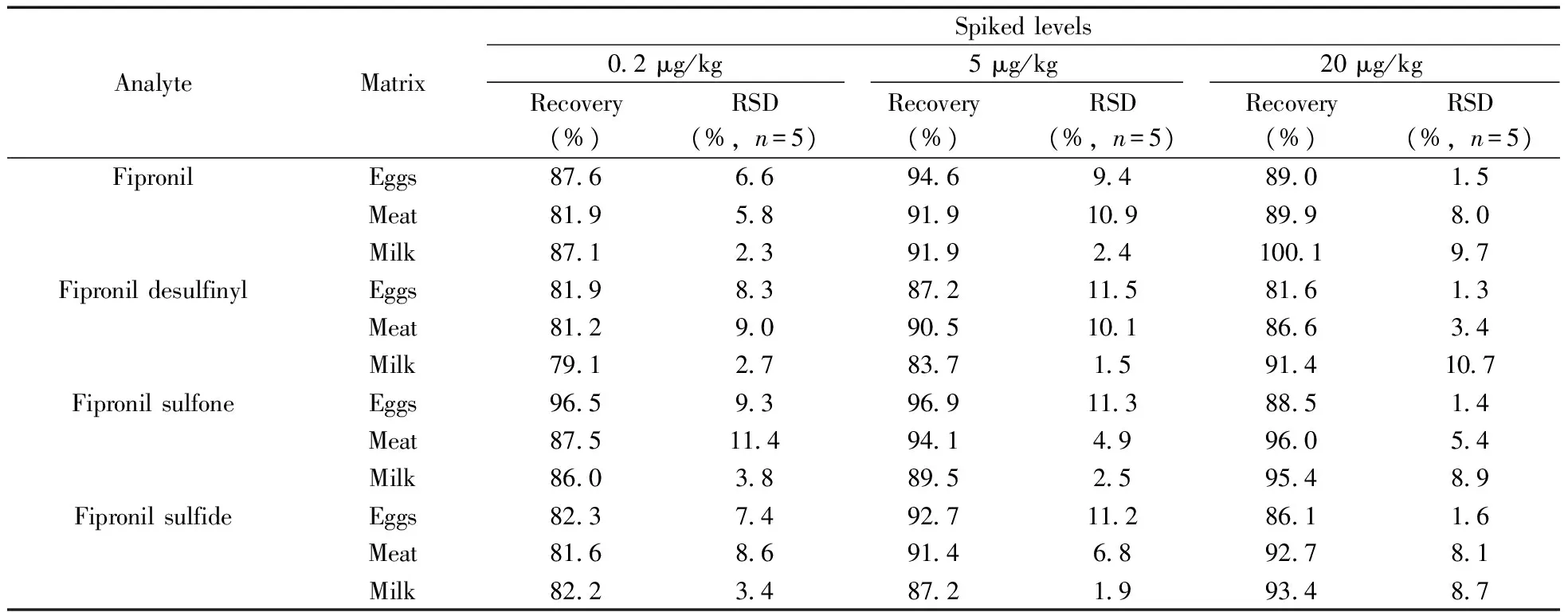
表3 不同加标水平下的回收率和相对标准偏差Table 3 Recovery and relative standard deviation of different spiked levels
2.6 实际样品的测定
采用本方法检测市售31份鸡蛋、3份鸡肉、6份猪肉、4份猪肝和5份牛奶等样品中的氟虫腈及其代谢物残留,结果显示,在鸡肉、猪肉、猪肝和牛奶样品中均未检测到氟虫腈及其代谢物,但有8份鸡蛋样品中检出氟虫腈砜,含量为0.13~1.1 μg/kg,低于欧盟所规定的最大残留限量。
3 结 论
本文采用超高效液相色谱-高分辨质谱技术,结合冷冻处理和分散固相萃取净化,首次建立了动物性食品中氟虫腈及其代谢物的分析方法,并应用于实际样品定性和定量的快速筛查分析。本方法具有样品前处理操作简单、价格低和基质效应低等特点,有利于快速监测动物性食品中氟虫腈及其代谢物的残留情况;高分辨质谱技术的PRM扫描模式与三重四极杆MRM模式相似,但由于其提供精确的碎片质量数,可进一步降低背景噪音的干扰,提高检测方法的灵敏度。
[1] Tingle C C D,Rother J A,Dewhurst C F,Lauer S,King W J.Rev.Environ.Contam.Toxicol.,2003,167(1):1-66.
[2] The German Federal Institute for Risk Assessment(BfR).Health Assessment of Individual Measurements of Fipronil Levels Detected in Foods of Animal Origin in Belgium.BfR Opinion No.016/2017 of 30 July 2017.
[3] Hurley P M,Hill R N,Whiting R L.Environ.HealthPersp.,1998,106(1):437-445.
[4] Zhang F F,Hong Y Q,Zhang X.Occup.Health.(张芳芳,洪雅青,张幸.职业与健康),2008,24(20):2211-2213.
[5] GB 2763-2016.National Food Safety Standard—Maximum Residue Limits for Pesticides in Food.National Standards of the People′s Republic of China(食品安全国家标准——食品中农药最大残留限量.中华人民共和国国家标准).
[6] Zhou Y,Xu D M,Chen D J,Zhang Z G,Zheng X H,Fang E H.Chin.J.Chromatogr.(周昱,徐敦明,陈达捷,张志刚,郑向华,方恩华.色谱),2011,29(7):656-661.
[7] Ramasubramanian T,Paramasivam M,Jayanthi R,Chandrasekaran S.FoodChem.,2014,150(1):408-413.
[8] Yan S Y,Li X H,Zhao E C,Jia C H.Chin.J.Anal.Lab.(闫思月,李兴海,赵尔成,贾春虹.分析试验室),2016,35(4):386-389.
[9] Bai B Q,Li M P,Zhang S W.FoodSci.(白宝清,李美萍,张生万.食品科学),2014,35(24):254-268.
[10] Lin T,Fan J L,Yang D S,Liu X Y,Shao J L,Li Y G,Liu H C.J.Instrum.Anal.(林涛,樊建麟,杨东顺,刘兴勇,邵金良,李彦刚,刘宏程.分析测试学报),2015,34(12):1360-1365.
[11] Du Y Y,Luo Y L,Wang J Q,Wu D M,Zhai Y Z,Yin X Y,Xu N S.Environ.Chem.(堵燕钰,罗漪涟,王洁琼,吴冬梅,翟云忠,殷雪琰,徐牛生.环境化学),2017,36(4):928-930.
[12] Liu Z Z,Qi P P,Wang X Y,Wang Z W,Xu X H,Chen W X,Wu L Y,Zhang H,Wang Q,Wang X Q.FoodChem.,2017,230(1):423-431.
[13] Zhang M Y,Bian K,Zhou T,Song X Q,Liu Q Y,Meng C Y,He L M.J.Chromatogr.B,2016,1014(1):31-36.
[14] FAO.Pesticide Residues in Food.Fipronil(202).2001:191-365.
[15] Sun B X,Guo D H,Ding Z P,Ji F,Dong J C,Yao J T.J.Instrum.Anal.(孙碧霞,郭德华,丁卓平,冀峰,董吉川,姚劲挺.分析测试学报),2010,29(10):1017-1024.
[16] Kaserzon S L,Heffernan A L,Thompson K,Mueller J F,Ramos M J G.Chemosphere,2017,182(1):656-664.
[17] Chen D W,Lü B,Zou J H,Yang X,Miao H,Zhao Y F.J.Instrum.Anal.(陈达炜,吕冰,邹建宏,杨欣,苗虹,赵云峰.分析测试学报),2014,33(12):1327-1333.
[18] Chen D W,Zhao Y F,Miao H,Wu Y N.J.Chromatogr.A,2014,1374(1):268-272.
[19] Chen D W,Zhao Y F,Miao H,Wu Y N.Talanta,2015,134(1):144-152.
[20] Chen D W,Miao H,Zou J H,Cao P,Ma N,Zhao Y F,Wu Y N.J.Agric.FoodChem.,2015,63(2):485-492.
[21] He M,Song D,Dong F S,Zheng Y Q.Environ.Chem.(贺敏,宋丹,董丰收,郑永权.环境化学),2016,35(5):925-932.
[22] Gong X M,Hua M M,Wang H T,Wang B J,Ma R H.J.Instrum.Anal.(宫小明,华萌萌,王洪涛,王炳军,马荣桧.分析测试学报),2017,36(7):897-901.
[23] Anastassiades M,Lehotay S J,Stajnbaher D,Schenck F J.J.AOACInt.,2003,86:412-431.
[24] Besil N,Cesio V,Heinzen H,Fernandez-Alba A R.J.Agric.FoodChem.,2017,65(23):4819-4829.
Determination of Fipronil and Its Metabolites in Animal Derived Foods by QuEChERSMethod Combined with Ultra Performance Liquid Chromatography-High-resolution Benchtop Q Exactive Mass Spectrometry
LÜ Bing1,YIN Shuai-xing2,CHEN Da-wei1*,FANG Cong-rong1,ZHOU Shuang1,ZHAO Yun-feng1
(1.China National Center for Food Safety Risk Assessment/Key Laboratory of Food Safety Risk Assessment,Ministry of Health,Beijing 100021,China;2.Shimadzu-GL Sciences(Shanghai)Laboratory Supplies Co.,Ltd.,Beijing 100027,China)
Based on cryogenic treatment and QuEChERS method,an ultra performance liquid chromatography-high-resolution benchtop Q Exactive mass spectrometry method for the determination of fipronil and its metabolites in animal derived foods was established.The samples were extracted with acetonitrile,and cleaned up by the cryogenic treatment and dispersive solid phase extraction(d-SPE)with the N-propylethlene diamine(PSA)and C18.The chromatographic analysis was performed on a BEH C18column with 0.1% acetic acid and acetonitrile as mobile phase by gradient elution,and the external standard calibration was used for quantification.The negative ion acquisition mode was applied,and the quantitative analysis was carried out by high resolution mass spectrometry using parallel reaction monitoring(PRM)mode.Fipronil and its metabolites showed good linearities in the concentration ranges of 0.02-2 μg/L and 0.2-20 μg/L,with the correlation coefficients(r2)above 0.992.For liquid or semi-liquid samples(such as milk and eggs)and the solid samples(such as chicken),the limits of quantification(LOQs)for 4 analytes were between 0.1 μg/kg and 0.2 μg/kg.At different spiked levels,the average recoveries of fipronil and its metabolites in eggs,chicken and milk were in the ranges of 81.6%-96.9%,81.2%-96.0% and 79.1%-100.1%,with the relative standard deviations(RSDs)of 1.3%-11.5%,3.4%-11.4% and 1.5%-10.7%,respectively.The method was simple,sensitive and accurate,and was suitable for the rapid screening and quantitative analysis of fipronil and its metabolites in animal derived foods,and the sensitivity of the method could meet the requirement for residual limitation of EU.
fipronil;animal derived food;QuEChERS;high-resolution mass spectrometry;metabolites
2017-08-22;
2017-08-30
国家食品安全风险评估中心高层次人才队伍建设523项目
*
陈达炜,博士,副研究员,研究方向:食品卫生,Tel:010-67779768,E-mail:chendw@cfsa.net.cn
10.3969/j.issn.1004-4957.2017.12.002
O657.7
A
1004-4957(2017)12-1424-07
猜你喜欢
现代临床医学(2022年4期)2022-09-29 07:36:10
食品安全导刊(2021年21期)2021-08-30 08:21:32
分析仪器(2019年3期)2019-06-18 08:38:58
农药科学与管理(2019年2期)2019-01-05 14:01:42
环球时报(2017-08-03)2017-08-03 11:55:02
化工科技(2016年6期)2016-06-06 01:54:24
灾害医学与救援(电子版)(2016年3期)2016-03-11 20:18:06
分析测试学报(2015年7期)2016-01-13 06:19:16
质谱学报(2015年5期)2015-03-01 03:18:37
当代畜禽养殖业(2014年12期)2014-02-27 08:00:13
