
钾钡共掺菲分子结构和电子特性的第一性原理研究
2017-12-25 06:26:53轩书科
物理学报 2017年23期
轩书科
(山东工艺美术学院公共课教学部,济南 250300)
钾钡共掺菲分子结构和电子特性的第一性原理研究
轩书科†
(山东工艺美术学院公共课教学部,济南 250300)
电子结构,第一性原理,有机超导体,芳香烃
1 引 言
超导研究一直是凝聚态物理领域的重要研究内容,发现和探寻新的超导材料是该研究的重要方向之一.2010年,日本Kubozono课题组在钾掺杂苉有机分子晶体中发现高达18 K的超导转变温度,这标志着新的有机超导体家族的诞生,将超导电性的研究拓展到具有π电子的芳烃分子晶体领域,并引起了广泛关注[1].
芳香烃分子的种类繁多,多个苯环以不同的方式相结合,具有元素构成简单、易于剪裁和修饰等优点.在芳烃分子晶体中通过插层或掺杂不同价态、不同数量的金属原子,可以形成新的复合材料,表现为有机导体或有机超导体[2,3].2011年6月,Kubozono课题组报道了金属钾掺杂的六苯并苯样品中的超导相变[3].同年10月,中国科学技术大学陈仙辉课题组报道了金属钾掺杂的菲晶体中超导转变温度Tc为5 K的超导相变[4];由磁化率测量得出菲分子上有0.2µB的局域磁矩,并指出这种超导体具有非常规超导电性.此后,该课题组相继报道了碱土金属Sr,Ba和稀土金属La,Sm掺杂的菲分子晶体的超导电性[5,6].2012年,中国科学院物理研究所陈根富课题组在钾掺杂的1,2:8,9-二苯并五苯分子晶体中实现了Tc为33 K的超导相变[7].2014年,北京高压科学研究中心陈晓嘉课题组报道了不同钾掺杂菲晶体中的超导,验证了陈仙辉课题组的结论[8].菲、苉、二苯并五苯分子中苯环按照折线形式排列,那么直线型分子的芳烃晶体掺杂后能否出现超导电性?中国科学院物理研究所靳常青课题组在钾掺杂的并五苯中发现了Tc为4.5 K的超导转变,突破了超导只能发生在直链分子晶体中的经验性结论[9].湖北大学黄忠兵课题组合成了钾掺杂对三联苯的超导样品[10],也有课题组尝试使用其他芳烃分子晶体合成超导材料,如六苯并苯、萘、蒽、并四苯、䓛、对三联苯.此外,在金(111)面、石墨等衬底上生长的钾掺杂六苯并苯和二萘品苯薄膜,其电性和磁性测量也有相关报道[11−13].
金属原子的掺杂浓度是合成芳烃类超导体的关键因素.在金属钾掺杂的苉和菲分子晶体中,金属原子与分子的化学计量比为3:1时,在部分样品中出现超导相变.在有些实验中,只有比例为3:1的样品中才出现超导[3],与过去Cs掺杂富勒烯实验中的比例一致.从电荷转移的角度看,当金属原子与分子的比例为3:1时,平均每个分子得到3个电子,其中两个电子占据分子的最低未占据轨道(lowest unoccupied molecular orbital,LUMO),第三个电子占据LUMO+1轨道,所以是半占据的.对于掺杂后的分子晶体而言,假定能带是分立的,由LUMO+1轨道形成的能带穿过费米能级,形成金属态.因此,在随后的实验和理论研究中,“三电子转移”受到重视[14−19],实验化学配比和理论模型大多符合每个分子得到三个电子.但是,由于芳烃晶体掺杂的实验样品合成十分困难,合成样品的质量参差不齐,对空气和水蒸气十分敏感,易于退化,限制了实验测量.目前,这类超导样品的成分、晶体结构等关键数据仍然是未知的,需要进一步进行理论和实验研究.
菲分子晶体的结构符合P21空间群,分子按照人字形交叉模式,在ab晶面内先排列成分子层,然后沿着c轴形成晶体.分子层中会形成网格或孔洞,每个孔洞由四个分子围成,孔洞的长度取决于分子长度.理论模拟和实验测量证明,金属原子在插层或掺杂后,倾向于占据分子层中的孔洞,而不是层与层之间的空隙[20−23].在芳烃分子晶体掺杂制备超导样品的过程中,研究人员尝试了不同的掺杂金属,比如碱金属钾、铷、铯,碱土金属钡、锶,稀土金属镧、钐.上述金属掺杂制备的样品都在实验中观测到了超导现象.但是在之前的理论和实验研究中,都是只掺入某种单一金属,两种或两种以上金属共掺杂的方案尚未提出.另外,三电子转移到每个分子是超导产生的一个有利条件[1,4,7,8,22],但有时受限于晶体结构,单纯的钾或钡掺杂不能满足这一条件,则需要同时掺杂两种金属原子.再者,金属共掺杂对电子结构的影响也是一个值得研究的课题.
本文从理论计算角度模拟了钾、钡共掺杂的电子结构,指出钾、钡共掺杂是在菲分子晶体中实现三电子转移到每个分子的一个可行方案,展示了钾、钡共掺杂的菲晶体的电子结构特征,发现有能带穿过费米能级,并具有较大的态密度.
2 模型构建与计算方法
2.1 晶体结构模型

图1 (网刊彩色)菲分子晶体结构 (a)晶体原胞;(b)晶体中分子层的排列;(c)分子层内的空隙;(d)掺杂金属的位置Fig.1.(color online)Crystal structure of phenanthrene:(a)Unit cell of phenanthrene crystal;(b)allignment of molecular layer in crystal;(c)interstitial space in molecular layer;(d)positions of doped metal atoms in phenanthrene crystal.
菲分子晶体的结构如图1所示,每个晶体原胞中包含两个分子,一个分子上的原子根据P21对称操作(x,y,z)→(−x,y+0.5,−z)和另一个分子上的原子对应.掺杂的金属原子进入分子层内的孔洞中.对于K1Ba1-菲而言,每个原胞内要掺入四个金属原子,它们的初始位置也符合上述对称操作.
2.2 理论计算方法
在电子结构计算中,我们采用基于密度泛函理论(DFT)的第一性原理平面波自洽场方法[16],原子间的相互作用由赝势来模拟.波函数展开采用平面波基矢,并用PBE(Perdew-Burke-Ernzerhof)型广义梯度近似来处理交换关联势部分[24],平面波的动能截断取500 eV.布里渊区中,k网格点的选取采用Monkhorst-Pack方案,网格尺寸为4×4×4,积分采用高斯展宽技术.主要计算用VASP(Viennaab-initioSimulation Package)软件包实现,采用的赝势取自VASP官方网站,选取的C,H,K,Ba原子的价电子组态为C(2s2p2),H(1s1),K(3s2p64s1),Ba(5s2p66s2),其他轨道电子视为芯电子.为了准确模拟分子晶体的结构,在计算中按照Langreth和Lundqvist建议的vdW-DF2方案考虑范德瓦尔斯力修正[25].
3 结果和分析
3.1 未掺杂菲分子晶体的理论模拟
首先进行纯菲的晶体结构模拟,从而检验所采用的计算方法和选取的计算参数是否可行.理论优化的纯菲的晶格参数如表1所示.
从表1可以看出,如果不计入范德瓦尔斯力,得到的晶格参数偏大,而且与实验值相差较大,尤其是a轴的值9.28 Å (1 Å =0.1 nm)远大于实验值8.46 Å;如果在计算中计入范德瓦尔斯力修正,得到的晶格参数与实验值几乎一致.由此说明,在这类材料的计算中,考虑范德瓦尔斯力修正十分必要.这也和分子晶体中分子间相互作用是一种弱的范德瓦尔斯力相符合.

表1 理论优化的菲晶体结构参数与实验值的比较Table 1.Comparison of the theoretically optimized structural parameters of pristine phenanthrene and the experimental ones.
3.2 掺杂剂的选择
在铁硒超导研究中,碱金属钾曾作为电子供体插层在铁硒层之间[27].在芳烃超导材料实验中,一价碱金属钾、铷、铯和二价碱土金属钡、锶都被选为掺杂剂.钾的离子半径为1.38 Å,钡的离子半径为1.35 Å,选择钾钡共掺杂可以保证掺杂后分子不发生明显的畸变和扭曲.这种平整的分子结构有利于芳烃分子π电子的离域性.
为了定量表示掺杂的难易程度,我们依据以下方法计算掺杂的形成能.计算表达式为

式中EFormation表示平均到每个掺杂金属原子上的形成能,EK1X1PHA表示图2所示一个金属菲复合物晶体原胞的能量,EPHA表示一个菲分子晶体原胞的能量,EK表示单个钾原子在金属钾中的能量,EX表示掺杂剂X(X=Ba,Sr,Ca)单个原子在金属块体中的能量.掺杂后的菲晶体K1X1-菲结构如图2所示,我们采用此结构计算形成能.(1)式中各个参量的具体数值列于表2中.计算得到K1Ba1-菲、K1Sr1-菲和K1Ca1-菲的形成能分别为−0.25,−0.13,−0.04 eV.这些形成能的数据进一步证明了钾钡共掺杂优于钾锶共掺杂和钾钙共掺杂.

表2 形成能计算中的参量值Table 2. The variables used in the formation energy calculation for K1Ba1-phenanthrene,K1Sr1-phenanthrene and K1Ca1-phenanthrene.
我们还计算了K2-菲和Ba2-菲的形成能,分别为−0.30 eV/K和−0.31 eV/Ba,略大于K1Ba1-菲的形成能−0.25 eV/atom.为粗略估计K1Ba1-菲相对于K2-菲和Ba2-菲的生成概率,由玻尔兹曼分布推导可得
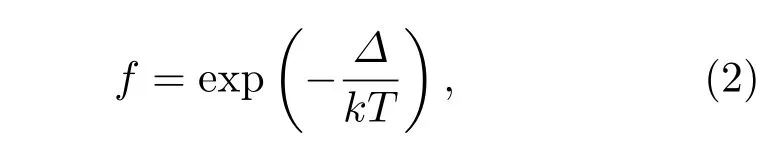
式中f为相对生成概率,Δ为能量差,实验制备样品的温度约为500 K,kT约为0.043 eV.因此,K1Ba1-菲相对于K2-菲的生成概率为
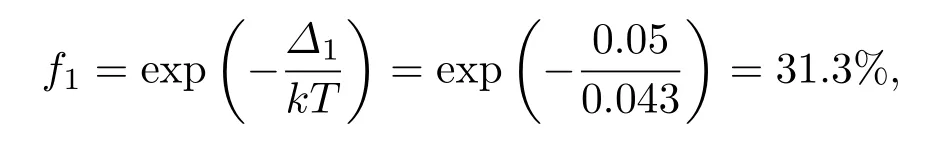
K1Ba1-菲相对于Ba2-菲的生成概率为

可以推断钾钡共掺杂制备K1Ba1-菲的方案在实验上是可行的.
3.3 钾钡共掺杂的菲分子晶体结构
最低能量态的选择过程中,我们考虑了三种情况.首先,计算钾或钡金属原子放在菲分子层之间的情况,发现体系能量太高.其次,将两个钾原子和两个钡原子分别放在两个孔洞中,此时金属原子排列不符合P21空间群对称操作(x,y,z)→(−x,y+0.5,−z).最后,当每个孔洞中都放置一个钾和一个钡原子,并且两个钾原子或钡原子都符合P21空间群对称性时,能量最低,如图2所示.
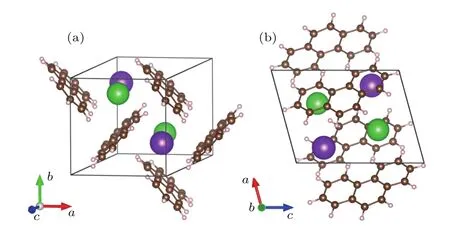
图2 (网刊彩色)钾钡共掺杂后的菲分子晶体结构(绿色和紫色原子分别代表钡原子和钾原子,它们均匀分布于分子层内) (a)晶体原胞正面图;(b)晶体原胞侧面图Fig.2. (color online)Crystal structure of K1Ba1-phenanthrene(green and purple balls represent barium and potassium atoms,respectively,and they are distributed uniformly in crystal):(a)Front view of a unit cell;(b)side view of a unit cell.
考虑到钾、钡原子间合理的间距,一个空隙中安放两个杂质原子是合适的.优化后的结构仍然保持P21群对称性,晶格参数分别为a=7.56 Å,b=7.73 Å,c=10.07 Å,β=105.28°. 由图2可以看出,由于金属原子的插入,掺杂后的晶体结构中分子间空隙变大,同一个原胞内的分子平面之间接近垂直关系.原胞内两个钡原子的z方向坐标分别为0.246c和0.754c;两个钾原子的z方向坐标分别为0.709c和0.291c.优化后掺杂的晶体结构如图2所示,从图中可以看出,菲分子在掺杂后仍然保持平整的结构,并没有发生明显扭曲和畸变.
3.4 钾钡共掺杂菲分子晶体的电子结构
计算钾钡共掺杂菲分子晶体的能带结构,如图3所示. 其中,倒易空间中k点路径选为Γ(000)→Z(0 0 0.5)→R(0 0.5 0.5)→F(0 0.5 0)→Γ(000)→X(0.5 0 0).从能带图发现两个明显的特征:1)各能带均较为平坦,局限于较窄的能量范围内,这对应于分子之间较弱的相互作用,由分子晶体的特点所决定;2)每两个能带组合在一起,形成一个能带对.
每个晶体原胞包含两个分子,这个能带对来自两个相同的分子轨道.如费米能级之下的能带对来自两个分子的LUMO,费米能级之上的能带对来自两个LUMO+1轨道,位于−2.8 eV处的能带对与分子的最高占据轨道(highest occupied molecular orbital,HOMO)对应.Γ(0 0 0)→Z(0 0 0.5)和R(0 0.5 0.5)→F(0 0.5 0)两个k路径平行于k空间的c∗轴,能量基本保持不变,对应于较弱的分子层间相互作用.Z(0 0 0.5)→R(0 0.5 0.5)平行于F(0 0.5 0)→Γ(0 0 0),Γ(0 0 0)→X(0.5 0 0)能量随着k的变化发生明显弥散,对应于空间中的b轴和a轴方向,说明引入掺杂的金属原子后,层内分子间、分子与杂质之间的相互作用加强.
通过Bader电荷分析可以检查金属原子上的电荷转移情况,每个钾原子失去0.8个电子,每个钡原子失去1.6个电子,这些电子转移到菲分子上.钾原子和钡原子的价态是+1和+2价.在钾钡共掺杂体系K1Ba1-菲中,金属原子为每个菲分子提供3个电子;而在单纯采用钾掺杂的体系K2-菲中,每个菲分子获得2个电子.与单纯采用钾掺杂相比,钾钡共掺杂时菲分子能获得更多的转移电子.理论和实验均证实每个分子获得3个电子是出现超导的有利因素[1,4,7,8,22].由于菲晶体中分子间空隙较小,容纳的杂质原子少,在菲晶体中要实现这种3电子转移,钾钡共掺杂是可行的措施之一.
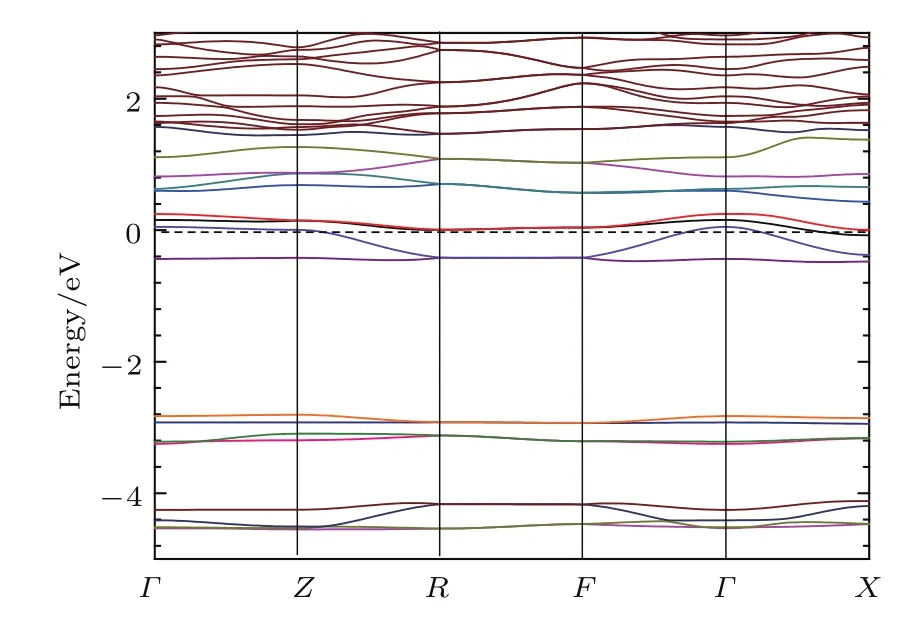
图3 (网刊彩色)钾钡共掺杂菲分子晶体的能带结构(费米能级设置为能量的零点)Fig.3. (color online)Band structure of K1Ba1-phenanthrene(the Fermi energy is set to zero).

图4 (网刊彩色)钾钡共掺杂菲分子晶体的总态密度和投影态密度 (a)总态密度和投影到钾钡原子上的态密度;(b)单个碳原子上的投影态密度Fig.4.(color online)Total densitiy of states and projected density of states of K1Ba1-phenanthrene:(a)Total density of states and its projection on K and Ba atoms;(b)the projected density of states on a carbon atom.
钾钡共掺杂菲晶体的态密度(density of states,DOS)如图4所示.从图4(a)可以看出,该体系是金属,费米能级处的态密度数值为17.3 eV−1.蓝线和红线分别表示钾原子和钡原子的电子态对总态密度的贡献,在图中所示能量范围内,它们的贡献很小,钾原子几乎没有贡献.图4(b)为单个碳原子2p轨道的投影态密度,其轮廓与图4(a)中的总态密度相似,在费米能级处碳原子2p轨道的电子态做出了主要贡献.如果进一步分析钡原子各个分轨道的贡献,我们发现其5d轨道而不是6s轨道对图中所示能量范围内的态密度做出了贡献.
3.5 掺杂前后电子结构的对比与分析
图5所示为掺杂前菲分子晶体的电子结构,包括总态密度和能带结构.对未掺杂的菲分子晶体,费米能级之上有2.8 eV的能隙,对应于菲分子的LUMO和HOMO之间的能隙.从图3和图4可以看出,掺杂K和Ba之后,该能隙位于费米能级之下−2.8—−0.5 eV之间,能隙宽度略减,变为2.3 eV.未掺杂的菲分子晶体是一个绝缘体,费米能级在价带顶,对应于菲分子HOMO形成的能带;掺杂K和Ba之后,费米能级越过能隙上移到原来的导带上,或者说占据了LUMO和LUMO+1轨道对应的能带,形成金属态.从掺杂K和Ba等金属的效果看,掺杂后的能带形状基本保持了分离的状态,不同能带对之间交叠很少,说明分子之间的相互作用仍然很弱.另一方面,K和Ba掺杂为体系增加了电荷,K和Ba的电子向菲分子转移,K和Ba掺杂属于电子型掺杂,提高了体系的费米能级.
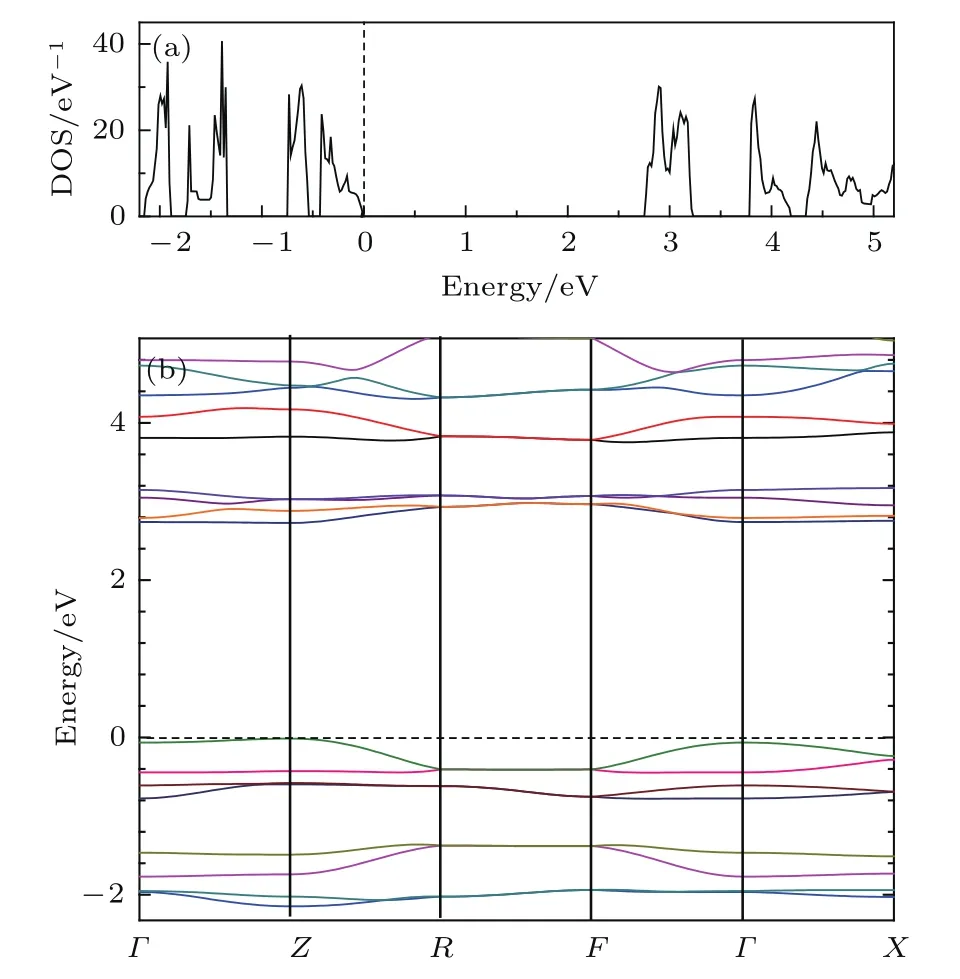
图5 (网刊彩色)菲分子晶体的总态密度和能带结构(a)总态密度(费米能级为0);(b)能带结构Fig.5.(color online)Total densitiy of states and band structure of pristine phenanthrene:(a)Total density of states(energy of Fermi level is set to 0);(b)structure.
4 结 论
提出在芳烃材料中采用不同价态金属共掺杂的想法,并从第一性原理计算的角度验证了此想法的可行性和必要性.首先通过原子半径的匹配和形成能比较了钾钡、钾钙、钾锶三种掺杂组合,得出钾钡组合是最佳掺杂方案.掺杂后,钾钡原子在分子层间均匀分布,但晶格参数相对于纯的菲晶体发生较大变化,a,b轴的长度近乎相等.能带显示分子层内的相互作用加强,但分子层间的相互作用依然较弱,费米能级处的能带来自LUMO和LUMO+1分子轨道.总态密度和投影态密度计算结果显示,C 2p轨道的电子态对费米能级附近的态密度做出了主要贡献.研究结果展示了钾钡共掺杂菲晶体的电子结构,并为芳烃超导体样品的制备提供了新的思路.
[1]Mitsuhashi R,Suzuki Y,Yamanari Y,Mitamura H,Kambe T,Ikeda N,Okamoto H,Fujiwara A,Yamaji M,Kawasaki N,Maniwa Y,Kubozono Y 2010Nature464 76
[2]Okazaki H,Jabuchi T,Wakita T,Kato T,Muraoka Y,Yokoya T 2013Phys.Rev.B88 245414
[3]Kubozono Y,Mitamura H,Lee X,He X,Yamanari Y,Takahashi Y,Suzuki Y,Kaji Y,Eguchi R,Akaike K,Kambe T,Okamoto H,Fujiwara A,Kato T,Kosugi T,Aoki H 2011Phys.Chem.Chem.Phys.13 16476
[4]Wang X F,Liu R H,Gui Z,Xie Y L,Yan Y J,Ying J J,Luo X G,Chen X H 2011Nat.Commun.2 507
[5]Wang X F,Yan Y J,Gui Z,Liu R H,Ying J J,Luo X G,Chen X H 2011Phys.Rev.B84 214523
[6]Wang X F,Luo X G,Ying J J,Xiang Z J,Zhang S L,Zhang R R,Zhang Y H,Yan Y J,Wang A F,Cheng P,Ye G J,Chen X H 2012J.Phys.Condens.Matt.24 345701
[7]Xue M,Cao T,Wang D,Wu Y,Yang H,Dong X,He J,Li F,Chen G F 2012Sci.Rep.2 389
[8]Huang Q W,Zhong G H,Zhang J,Zhao X M,Zhang C,Lin H Q,Chen X J 2014J.Chem.Phys.140 114301
[9]Nakagawa T,Yuan Z,Zhang J,Yusenko K V,Drathen C,Liu Q,Margadonna S,Jin C 2016J.Phys.Condens.Matt.28 484001
[10]Gao Y,Wang R S,Wu X L,Cheng J,Deng T G,Yan X W,Huang Z B 2016Acta Phys.Sin.65 077402(in Chinese)[高云,王仁树,邬小林,程佳,邓天郭,闫循旺,黄忠兵2016物理学报65 077402]
[11]Wu X,Xu C,Wang K,Xiao X 2016J.Phys.Chem.C120 15446
[12]Phan Q T N,Heguri S,Tamura H,Nakano T,Nozue Y,Tanigaki K 2016Phys.Rev.B93 075130
[13]Kambe T,Nishiyama S,Nguyen H L T,Terao T,Izumi M,Sakai Y,Zheng L,Goto H,Itoh Y,Onji T,Kobayashi T C,Sugino H,Gohda S,Okamoto H,Kubozono Y 2016J.Phys.Condens.Matt.28 444001
[14]Kosugi T,Miyake T,Ishibashi S,Arita R,Aoki H 2011Phys.Rev.B84 214506
[15]de Andres P L,Guijarro A,Vergés J A 2011Phys.Rev.B83 245113
[16]Giovannetti G,Capone M 2011Phys.Rev.B83 134508[17]Naghavi S S,Fabrizio M,Qin T,Tosatti E 2013Phys.Rev.B88 115106
[18]Zhong G,Huang Z,Lin H 2014IEEE Trans.Magn.50 1700103
[19]Yan X W,Huang Z,Lin H Q 2013J.Chem.Phys.139 204709
[20]Yan X W,Huang Z,Lin H Q 2014J.Chem.Phys.141 224501
[21]Yan X W,Zhang C,Zhong G,Ma D,Gao M 2016J.Mater.Chem.C4 11566
[22]Dutta T,Mazumdar S 2014Phys.Rev.B89 245129
[23]Yan X W,Wang Y,Gao M,Ma D,Huang Z 2016J.Phys.Chem.C120 22565
[24]Perdew J P,Burke K,Ernzerhof M 1996Phys.Rev.Lett.77 3865
[25]Klimeš J,Bowler D R,Michaelides A 2011Phys.Rev.B83 195131
[26]Kay M I,Okaya Y,Cox D E 1971Acta Cryst.B27 26
[27]Guo J,Sun L L 2015Acta Phys.Sin.64 217406(in Chinese)[郭静,孙力玲 2015物理学报 64 217406]
First principles studies on molecular structure and electronic properties of K-and Ba-codoped phenanthrene
Xuan Shu-Ke†
(School of Common Courses,Shandong University of Art and Design,Ji’nan 250300,China)
10 March 2017;revised manuscript
25 April 2017)
The superconductivity has always been one of the important topics in condensed matter physics.Recently,the discovery of superconductivity in potassium-doped picene have opened the way to a new class of organic superconductor,and at the same time metal-doped aromatic hydrocarbons have attracted great interest of researchers in investigating their physical and chemical properties.In this paper,according to the plane wave and pseudopotential method in the framework of density functional theory,we systematically study the structural and electronic properties of the K/Bacodoped phenanthrene,including the atomic structure,band structure,density of states,formation energy,and charge transfer between dopant and phenanthrene molecule,and three meaningful conclusions have been drawn as follows.At first,the van der Waals interaction is found to play an important role in determining the atomic structure of metal-doped molecular solid,so it is necessary to include the interactions in these calculations.Secondly,due to the similarity in ionic radius,the combination of K and Ba is the favorable scheme for multiple-metal codoped phenanthrene crystal compared with K/Ca and K/Sr codoping schemes.From the viewpoint of formation energy,K1Ba1-phenanthrene has a bigger formation energy(−0.25 eV)per doped metal atom than K1Sr1-phenanthrene(−0.13 eV)and K1Ca1-phenanthrene(−0.04 eV).Thirdly,in order to realize the−3 valent state of phenanthrene molecule in K/Ba-codoped phenanthrene,the codoping of monovalent and bivalent metals is the only viable option due to the narrow interstitial space in molecular crystal.The bands crossing the Fermi level are from the lowest unoccupied molecular orbital(LUMO)and LUMO+1 orbital,resulting in the metallic state of K1Ba1-phenanthrene.The large density of states at the Fermi level is 17.3 eV−1,and these electronic states are mainly from C 2p orbitals and a little contribution from Ba 5d orbitals.Our studies present the electronic structure of K1Ba1-phenanthrene and suggest that K/Ba-codoping is a rational scheme to synthesize the superconductive sample,which provides a new route to the exploration of the promising superconductivity in metal-doped aromatic hydrocarbons.
electronic structure, first principles,organic superconductor,aromatic hydrocarbon
PACS:74.70.Kn,74.20.Pq,61.48.–cDOI:10.7498/aps.66.237401
†Corresponding author.E-mail:shuke158@126.com
(2017年3月10日收到;2017年4月25日收到修改稿)
采用基于密度泛函理论的平面波赝势法,对钾钡共掺杂情形下菲分子晶体的结构、能带、电子态密度、形成能、电荷转移等电子特性进行系统的研究.通过对比计入范德瓦尔斯力作用前后晶体结构的差异,说明计算中包含范德瓦尔斯力修正的重要性.从形成能的角度证明了共掺杂的可行性和稳定性.在钾钙、钾锶、钾钡等共掺杂元素组合中,K1Ba1-菲中平均每个金属原子的形成能为−0.25 eV,远大于K1Sr1-菲和K1Ca1-菲中的−0.13和−0.04 eV,钾钡共掺杂是最合理的方案.只有单双价金属共掺杂,才能使分子呈现负三价.此时,菲分子最低未占据轨道(LUMO)和LUMO+1轨道组成的能带正好位于费米能级处,K1Ba1-菲呈现金属性.费米能级处的态密度为17.3 eV−1,电子态主要来自于碳原子的2p轨道,钡原子的5d轨道也有少许贡献.从理论模拟的角度研究了K1Ba1-菲的晶体结构和电子特性,在已有实验和理论研究尚未涉及共掺杂的背景下,提出了不同价态金属共掺杂方案,为制备芳烃有机超导体样品和调制体系电子结构提供了新的研究思路.
10.7498/aps.66.237401
†通信作者.E-mail:shuke158@126.com
猜你喜欢
杭州(2023年3期)2023-04-03 07:22:04
小天使·聪聪画刊(2021年2期)2021-09-10 07:22:44
汽车零部件(2020年10期)2020-11-09 03:41:42
复旦学报(医学版)(2020年3期)2020-06-18 07:36:52
原子与分子物理学报(2020年5期)2020-03-17 07:00:00
数学物理学报(2019年6期)2020-01-13 06:08:24
发明与创新·小学生(2019年12期)2019-12-05 06:02:28
发明与创新(2019年47期)2019-11-21 01:16:18
汉语世界(The World of Chinese)(2019年6期)2019-09-10 07:22:44
深圳大学学报(理工版)(2015年5期)2015-02-28 16:21:26
