
苯并异硒唑酮类化合物的合成研究进展
2017-12-19 06:11阮奔放
浙江化工 2017年11期
张 正,侯 卫,阮奔放
(浙江工业大学药学院,浙江 杭州 310014)
苯并异硒唑酮类化合物的合成研究进展
张 正,侯 卫,阮奔放*
(浙江工业大学药学院,浙江 杭州 310014)
苯并异硒唑酮类化合物是一类重要的具有抗肿瘤、抗氧化和抗炎等生物活性和低毒性化合物,因此受到广泛关注,但目前苯并异硒唑酮类化合物合成方法不多,而且效果也不好。本文以起始原料分类,对苯并异硒唑酮类化合物的合成路线进行综述。
苯并异硒唑酮;合成;综述
0 前言
苯并异硒唑酮类化合物最早在上世纪20年代就被发现,但直至最近十几年它才因被发现具有非常好的谷胱甘肽过氧化物酶(GSH-Px)抗氧化活性而被广泛深入研究,其抗肿瘤和抗微生物活性也逐渐引起了人们的重视。如依布硒啉被发现具有抗肿瘤和抗微生物等活性,可以通过对硫氧环蛋白还原酶活性的抑制,从而实现药效作用[1],在进一步对依布硒啉的生物活性研究中发现依布硒啉同时具有优秀的谷氨酰胺酶抑制活性[2]。曾慧慧等[3]发现了创新抗肿瘤药物乙烷硒啉,目前已进入二期临床研究。陈宝泉等[4]将苯并异硒唑酮分子与1,3,4-噻二唑进行接枝,合成了一系列新化合物,在生物活性测试中发现对MCF-7、SSMC-7721和A549这三种癌细胞均有增殖抑制效果,且表现出一定的选择性。
苯并异硒唑酮类化合物由于其优秀的生物活性,在医药领域和药理学领域上备受瞩目,然而对其合成方法的研究却相对较少,这可能是由于硒元素作为氧族元素的一员有较大的体积并存在空的d轨道,这增加了与较小体积的氮元素的弱轨道重叠几率,导致了与氮原子形成的成键分子轨道较弱,容易断裂,故氮-硒键的形成比较困难[5]。
本文以起始原料分类,对苯并异硒唑酮类化合物的合成方法进行综述和评价。
1 合成方法
1.1 以邻氨基苯甲酸为原料
Welter等人[6]报道了以邻氨基苯甲酸为原料,与亚硝酸钠在酸性条件下形成重氮盐,再与二硒化钠发生亲核取代反应生成联硒中间体,而后在氯化亚砜中以DMF为催化剂得到2-硒氯苯甲酰氯,最终与胺缩合环化合成了一系列苯并异硒唑酮类化合物,反应总产率42%~55%(Scheme 1)。这也是使用最为广泛的苯并异硒唑酮类化合物的合成方法之一,该方法具有底物适应范围广泛、安全性高等优点,但反应步骤较多,反应时间较长。
1.2 以苯甲酰胺类化合物为原料
Engman等人[7]报道了以苯甲酰胺类化合物为原料,通过正丁基锂进行邻位锂化反应,并与硒粉在冰浴下反应,将硒原子插入碳-锂键之间,最后尝试了 Br2、I2、FeCl3、CuBr2等氧化剂将其环合得到产品,研究发现CuBr2的环化效果最好,总产率约 50%(Scheme 2)。 Oppenheimer等人[8]对此方法进行了改进,在第一步反应中以二异丙基氨基锂和正丁基锂作为锂化试剂在0℃下反应,使总产率提升至68%(Scheme 2)。Zade等人[9]尝试直接使用SeCl2对邻位锂化中间体进行环合,最终取得成功并缩短了反应步骤,但产率仅有40%(Scheme 2)。本方法无需对反应中间体进行分离纯化即可直接得到目标产物,操作简便,反应时间短。

Scheme 1

Scheme 2
Fong等人[10]基于 Engman等人[7]的部分工作上,先将苯甲酰胺类化合物邻位锂化,再与硒粉反应,反应结束后加入铁氰化钾水溶液即可分离出联硒中间体,联硒化合物通过二叔丁基过氧化物或过氧化苯甲酰以及氯苯发生分子内均裂取代,最终得到苯并异硒唑酮类化合物,总产率46%~51%(Scheme 3)。
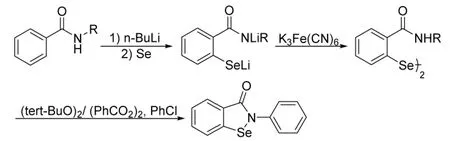
Scheme 3
Fong等人[11]同样以苯甲酰胺类化合物为原料,经邻位锂化并与二苄基二硒醚反应,再与光气室温下搅拌24 h转化为对应的氯化亚胺,不经分离纯化直接与1-羟基吡啶硫酮钠盐生成PTOC(pyridine-2-thioneoxycarbonyl)酰亚胺,在 200 W低压汞灯照射下即可得到目标产物,反应总产率36%~58%(Scheme 4)。

Scheme 4
1.3 以邻位卤代的苯甲酰胺类化合物为原料
Balkrishna等人[12]以2-卤代苯基酰胺类化合物为原料,DMF为溶剂,在110℃下加入邻菲罗啉、硒粉、碳酸钾,经碘化亚铜催化一步生成终产品,反应产率最高可达96%(Scheme 5)。其中原料中取代基为碘基时反应速度最快,取代基为溴基则需延长反应时间,但收率基本相当。本方法步骤短,实验操作简便,有一定的底物适应范围,反应条件对羟基、醚、酰胺、硝基、氟和氯等基团均较为稳定,但部分原料成本较高,且后处理时产物难以与催化剂分离。

Scheme 5
Pacuta等人[13]以2-碘代苯基酰胺类化合物为原料,与二硒化钠反应,通过一步反应即可生成苯并异硒唑酮类化合物,产率69%。他们随后尝试了使用二硒化钾和二硒化锂作为反应物,分别提升反应产率至75%和91%。后续实验表明,若为2-氯代原料,则无法发生反应;若为2-溴代原料,则反应产率大大降低至21%(Scheme 6)。
Singh等人[14]以2-溴代苯基酰胺类化合物为原料,在乙醇中与BuSeNa发生亲核取代反应,再通过溴素诱导环化得到产品,总产率38%~55%(Scheme 7)。

Scheme 6

Scheme 7
1.4 以邻位溴代的苯甲酸类化合物为原料
Lambert等人[15]以邻位溴代的苯甲酸类化合物与CH3SeH发生亲核取代反应生成2-甲基硒基苯甲酸,产率良好。再向得到的2-甲基硒基苯甲酸中加入氯化亚砜、催化量的DMF和胺,最终得到目标产物,产率最高可达78%(Scheme 8)。

Scheme 8
2 结论
苯并异硒唑酮类化合物由于其优秀的生物活性被广泛的应用于药物合成中。本文综述了已报道的一些苯并异硒唑酮类化合物的合成方法,各条路线在底物适应范围、反应时间、反应步骤等方面各有优劣,有待进一步的优化、改进。
[1]Luo Z,Sheng J,Sun Y,et al.Synthesis and evaluation of multi-target-directed ligands against Alzheimer’s disease based on the fusion of donepezil and ebselen[J].Journal of medicinal chemistry, 2013, 56(22): 9089-9099.
[2] Zhu M,Fang J,Zhang J, et al.Biomolecular interaction assays identified dual inhibitors of glutaminase and glutamate dehydrogenase that disrupt mitochondrial function and prevent growth of cancer cells[J].Analytical Chemistry, 2017, 89(3): 1689-1696.
[3] He J,Li D,Xiong K,et al.Inhibition of thioredoxin reductase by a novel series of bis-1,2-benzisoselenazol-3(2H)-ones: Organoselenium compounds for cancer therapy[J].Bioorganic&medicinal chemistry, 2012, 20(12):3816-3827.
[4] 史艳萍,陈宝泉,麻静,等.2-(2-取代-1,3,4-噻二唑-5-基)-苯并异硒唑-3(2H)-酮衍生物的合成及体外抗癌活性[J].化学学报, 2011,69(21):2561-2566.
[5]Saeed A,Larik F,Channar P.Synthetic approaches to the multifunctional drug ebselen and analogs:past and present[J].Mini-Reviews in Organic Chemistry, 2016, 13(4):312-324.
[6] Welter A, Christiaens L.New benzisoselenazolones, process for producing the same and pharmaceutical preparations containing the same:EP, 0044453[P].1982-01-27.
[7] Engman L,Hallberg A.Expedient synthesis of ebselen and related compounds[J].The Journal of Organic Chemistry, 1989, 54(12): 2964-2966.
[8] Oppenheimer J, Silks L A.Synthesis of 2-phenyl-1, 2-benziso[77Se]selenazol-3(2H)-one: “Ebselen”[J].Journal of Labelled Compounds and Radiopharmaceuticals,1996,38(3): 281-284.
[9] Zade S S, Panda S, Singh H B, et al.Synthesis of diaryl selenides using the in situ reagent SeCl2[J].Tetrahedron letters, 2005, 46(4): 665-669.
[10]Fong M C, Schiesser C H.Reactions of 2, 2′-diselenobis(N-alkylbenzamides) with peroxides: A free-radical synthesis of Ebselen and related analogues[J].Tetrahedron letters, 1995, 36(40): 7329-7332.
[11]Fong M C,Schiesser C H.Intramolecular homolytic substitution with amidyl radicals:a free-radical synthesis of Ebselen and related analogues[J].The Journal of Organic Chemistry, 1997, 62(10): 3103-3108.
[12]Balkrishna S J,Bhakuni B S, Chopra D, et al.Cu-catalyzed efficient synthetic methodology for Ebselen and related Se-N heterocycles[J].Organic letters, 2010, 12(23): 5394-5397.
[13]Pacuta A J,′Scianowski J,Aleksandrzak K B.Highly efficient synthesis and antioxidant capacity of N-substituted benzisoselenazol-3 (2 H)-ones[J].RSC Advances, 2014,4(90): 48959-48962.
[14]Singh V P, Poon J, Yan J, et al.Nitro-, azo-and amino derivatives of Ebselen: Synthesis, structure and cytoprotective effects[J].The Journal of Organic Chemistry, 2017,82(1): 313-321.
[15]Lambert C,Christiaens L.Stabilisation of condensed selenium heterocycles by a nitro group in the ortho-position to the chalcogen[J].Tetrahedron, 1991, 47(43): 9053-9060.
The Chemical Syntheses of Benzisoselenazolones
ZHANG Zheng, HOU Wei, RUAN Ben-fang*
(College of Pharmaceutical Science,Zhejiang University of Technology,Hangzhou,Zhejiang 310014,China)
Benzisoselenazolones are important compounds due to their anti-tumor,anti-oxidant,antiinflammatory activities and low toxicity.However,general and efficient synthetic methods remains rare.Here,we reviewed the reported methods based on the difference in starting materials.
benzisoselenazolones;chemical synthesis;review
1006-4184(2017)11-0020-04
2017-04-09
张正(1993-),男,硕士研究生,主要从事药物合成研究。
* 通讯作者:阮奔放,E-mail:ruanbf@zjut.edu.cn。
猜你喜欢
农业与技术(2021年3期)2021-12-06
世界科学技术-中医药现代化(2021年7期)2021-11-04
右江民族医学院学报(2021年4期)2021-09-18
现代农药(2021年2期)2021-05-07
科学大观园(2019年23期)2019-09-10
农药科学与管理(2019年10期)2019-04-20
天然产物研究与开发(2018年7期)2018-08-21
中国调味品(2017年2期)2017-03-20
百科知识(2016年22期)2016-12-24
百科知识(2016年16期)2016-10-29
