
1,3,5,7-四甲基氟硼二吡咯甲川的合成、晶体结构及密度泛函理论研究
2017-12-13 10:51段海潮易平贵于贤勇李筱芳
无机化学学报 2017年12期
段海潮 易平贵*, 于贤勇 李筱芳 刘 武 苏 剑
1,3,5,7-四甲基氟硼二吡咯甲川的合成、晶体结构及密度泛函理论研究
段海潮1易平贵*,1于贤勇1李筱芳1刘 武1苏 剑2
(1湖南科技大学化学化工学院,理论有机化学与功能分子教育部重点实验室,分子构效关系湖南省普通高校重点实验室,湘潭 411201)
(2南京大学化学化工学院,南京 210023)
以2,4-二甲基吡咯、原甲酸三乙酯以及对甲苯磺酸为原料,无溶剂下合成了1,3,5,7-四甲基对甲苯磺酸二吡咯甲川(T1);以T1、三氟化硼乙醚以及三乙胺为原料,无溶剂下合成了1,3,5,7-四甲基氟硼二吡咯甲川(F1)。利用1H NMR和X射线单晶衍射对其进行了表征。结果表明:T1属于单斜晶系,P21/c空间群,晶胞参数为a=0.777 3(3)nm,b=1.518 5(5)nm,c=1.612 2(5)nm,β=91.923(12)°,V=1.901 9(11)nm3,Z=4;F1 属于单斜晶系,P21/n 空间群,晶胞参数为 a=0.775 02(1)nm,b=1.444 20(3)nm,c=1.174 35(2)nm,β=107.779 5(9)°,V=1.251 65(4)nm3,Z=4。在4种不同溶剂中,测定了T1的紫外可见光谱、F1的紫外可见光谱和稳态荧光光谱。将密度泛函计算与前线轨道理论相结合,研究了T1和F1可能的衍生方式。
1,3,5,7-四甲基氟硼二吡咯甲川;核磁共振;晶体结构;荧光光谱;密度泛函理论
0 引 言
与传统染料相比,氟硼二吡咯甲川(Scheme 1)类荧光染料大多具有优异的光化学物理性能[1]。该类染料作为一种常见的近红外荧光染料,具有摩尔消光系数大、荧光量子产率高、光稳定性好、结构易于修饰、不易受环境溶液pH值影响等优点,已成为研究的热点[2]。由于其优良的光物理特性,氟硼二吡咯甲川荧光染料被广泛应用于荧光标记、荧光探针、电致发光器件如可调的激光器件、固态太阳能集线器、荧光开关等领域[3]。
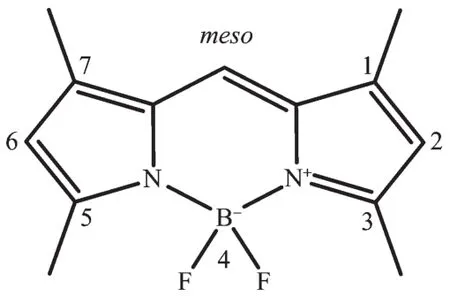
Scheme 1 Structure of 1,3,5,7-tetramethyl-4,4-difluoro-4-bora-3a,4a-diaza-s-indacene
近50年来,国内外研究工作者对这类化合物进行了深入的研究。1968年,此类荧光染料首次被Treibs和Kreuzer发现。1985年,Worries和Koek等首次在2位引入了磺酸基;此项研究为该染料广泛应用于生物分析领域开辟了新的道路,使人们认识到了此类染料潜在的应用价值[4]。吡咯上没有取代基[5]的和吡咯上有取代基[6]的该类化合物均有报导。为增加激发和发射波长以及促进该类材料在离子检测方面的应用,研究人员倾向于在meso位[7]、2,6位[8]、3,5 位[9]以及 4 位[10]实现衍生。
先前虽有关于1,3,5,7-四甲基氟硼二吡咯甲川的报导,但研究主要集中在合成和荧光方面,鲜见晶体结构和理论方面的研究。鉴于理论对实验的解释和预测方面具有重要意义,本文在合成以原甲酸三乙酯为原料的1,3,5,7-四甲基氟硼二吡咯甲川的基础上,利用核磁共振技术和单晶衍射技术对1,3,5,7-四甲基对甲苯磺酸二吡咯甲川 (以下简称T1) 和 1,3,5,7-四甲基氟硼二吡咯甲川 (以下简称F1)的分子结构进行了表征,结合密度泛函计算研究了2种化合物的反应性,并合理地解释了实验现象。
1 实验部分
1.1 仪器和试剂
所用试剂均为市售分析纯,未经进一步纯化。2,4-二甲基吡咯购于天津希恩思生化科技有限公司,原甲酸三乙酯购于基丽化学技术(上海)有限公司,对甲苯磺酸购于国药集团化学试剂有限公司经干燥后使用,三乙胺购于西陇化工股份有限公司,三氟化硼乙醚购于山东西亚化学股份有限公司,水为自制二次蒸馏水。
UV-Vis采用日本岛津公司的SHIMADZU UV-2501PC紫外光谱仪测定,稳态荧光采用日本岛津公司的Shimadzu RF-5301 PC荧光光谱仪测定,NMR采用瑞士Bruker有限公司的AV 500 MHz核磁共振仪测定,熔点采用上海精密科学仪器有限公司的WRS-1B数字熔点仪测定,高分辨质谱采用美国Agilent公司的6120 LC/MS型质谱仪测定。
1.2 1,3,5,7-四甲基氟硼二吡咯甲川的合成
1,3,5,7-四甲基氟硼二吡咯的合成参照文献[4]进行(Scheme 2)。向25 mL圆底烧瓶中依次加入对甲苯磺酸 (0.344 g,2 mmol),2,4-二甲基吡咯(415 μL,4 mmol),然后缓慢滴加原甲酸三乙酯(900 μL),并同时搅拌,滴加完毕后继续搅拌1 h。加入5 mL乙酸乙酯淬灭,减压抽滤,并用5 mL乙酸乙酯和5 mL石油醚各洗涤1次,50℃真空干燥得红色固体。将干燥过的红色固体装入100 mL茄形瓶中,向瓶中加入6 mL三氟化硼乙醚,磁力搅拌,然后缓慢滴加3 mL三乙胺,滴加完毕后继续搅拌1 h。加入20 mL二氯甲烷稀释,用50 mL二次蒸馏水分解余下的三氟化硼乙醚,分液,再用二次蒸馏水(50 mL×3)洗涤有机层,将有机层液体旋干,然后柱层析分离(V乙酸乙酯∶V石油醚=1∶25),收集红色液体(TLC 上有黄色荧光点)。将收集到的红色液体旋干后,50℃真空干燥得红色固体0.357 g,产率72%)。

Scheme 2
中间产物 T1:1H NMR(500 MHz,CDCl3):δ 12.79(s,2H,-NH),7.86(d,J=8.0 Hz,2H),7.19(d,J=8.0 Hz,2H),7.05(s,1H),6.14(s,2H),2.49(s,6H),2.36(s,3H),2.34(s,6H)。 m.p.194~195 ℃。 T1 为离子化合物,由两部分组成,分别是二吡咯甲川阳离子(cation,简称 C)、对甲苯磺酸根阴离子(anion,简称A)。 ESI-HRMS(C20H24N2O3S,CA),m/z:201.138 4[C]+,573.288 5[C+CA]+。
最终产物 F1:1H NMR(500 MHz,CDCl3):δ 7.04(s,1H),6.05(s,2H),2.53(s,6H),2.25(s,6H)。 m.p.209~212 ℃。ESI-HRMS(C13H15BF2N2),m/z:249.136 8[M+H]+。
1.3 晶体结构测定
晶体衍射数据在Bruker Smart APEX 1000 CCD衍射仪上,采用石墨单色化Mo Kα射线(λ=0.071 073 nm)在296 K下运用φ-ω扫描技术收集。收集的数据通过SAINTPLUS程序还原,SADABS做吸收校正[11-13];晶体结构用SHELXS-97程序由直接法解出[14-16]并用SHELXL-97程序[17-19]对所有非氢原子坐标和各向异性温度因子进行了全矩阵最小二乘法精修。有关晶体的详细信息列于表1和表2中。
CCDC:1578792,T1;1578790,F1。
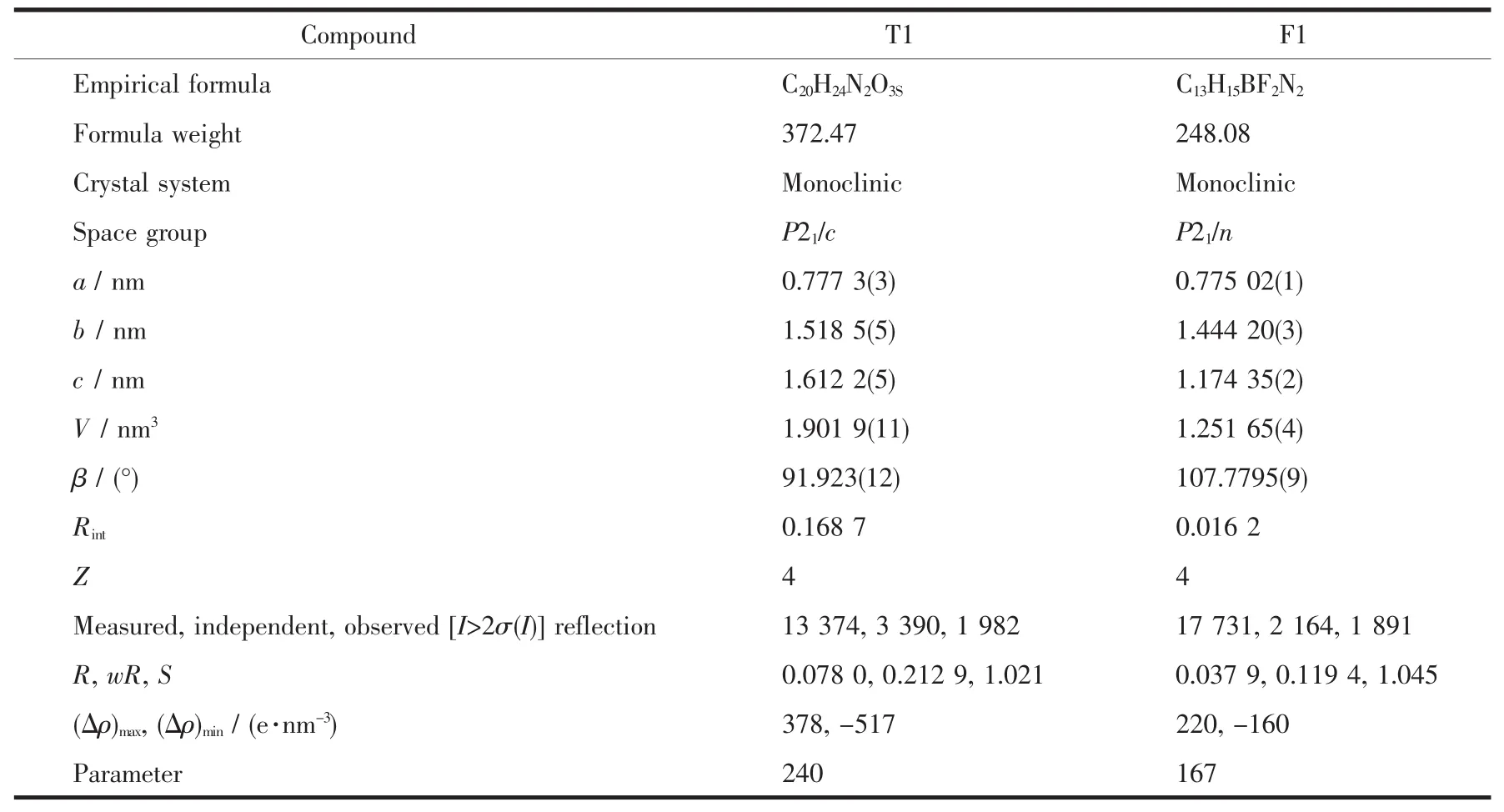
表1 T1和F1的结构精修参数Table 1 Structure refinement parameters for T1 and F1

表 2 T1 和 F1 晶体的部分键长(nm)、键角(°)及二面角(°)Table 2 Selected bond lengths(nm),bond angles(°)and dihedral angles(°)for T1 and F1
1.4 计算方法
采用密度泛函理论 (DFT)的计算方法,拟在B3LYP/6-31G(d)水平上,对气相条件下T1和F1进行全优化,通过振动频率分析确认其稳定性。计算溶剂作用时,在B3LYP/6-31G(d)水平上[20-24]结合PCM模型[25-28]对其基态进行全优化。全部计算在Gaussian 09软件包[29]上完成。
2 结果与讨论
2.1 晶体结构
2.1.1 中间产物T1
T1的结构如图1所示。图1中,2个吡咯环构成的二面角约为2°左右,接近于共平面。C5-C6和C6-C9键长分别为0.139 3(7)、0.137 5(7)nm,均接近于共轭环的双键键长0.138 nm。C5-C6-C9键角为134.2(4)°,接近于平面正八边形内角135°。上述结果可能是由于C6与2个吡咯环形成了较好的共轭,并且与 C5、C9、N1、N2、O1 以及 2 个氢原子形成了类似于平面八元环的结构。
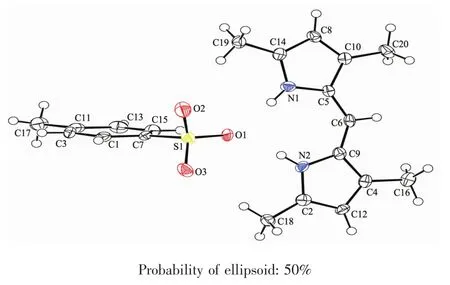
图1 T1的分子结构Fig.1 Molecular structure of T1
2.1.2 最终产物F1
F1 的结构如图 2 所示。图 2 表明,1,3,5,7-四甲基氟硼二吡咯已被成功合成,并有着较好的平面结构。其π电子数为10,满足4n+2规则,因此由休克尔规则可知,该化合物具有芳香性,易发生亲电取代反应。
2.2 紫外可见光谱
图3和图4分别为T1及F1的紫外可见光谱。测量时,T1 和 F1 的浓度均为 2×10-5mol·L-1。T1 在环己烷、乙醇、N,N-二甲基甲酰胺以及水中的最大吸收波长分别为 468、465、467 和 462 nm;其中,乙醇中的T1对465 nm波长光的响应最大,此时该化合物的摩尔消光系数为 9.987×104L·mol-1·cm-1。 F1 在环己烷、乙醇、N,N-二甲基甲酰胺以及水中的最大吸收波长分别为 508、503、503 和 497 nm;其中,环己烷中的F1对508 nm波长光的响应最大,此时该化合物的摩尔消光系数为 1.128 5×105L·mol-1·cm-1。
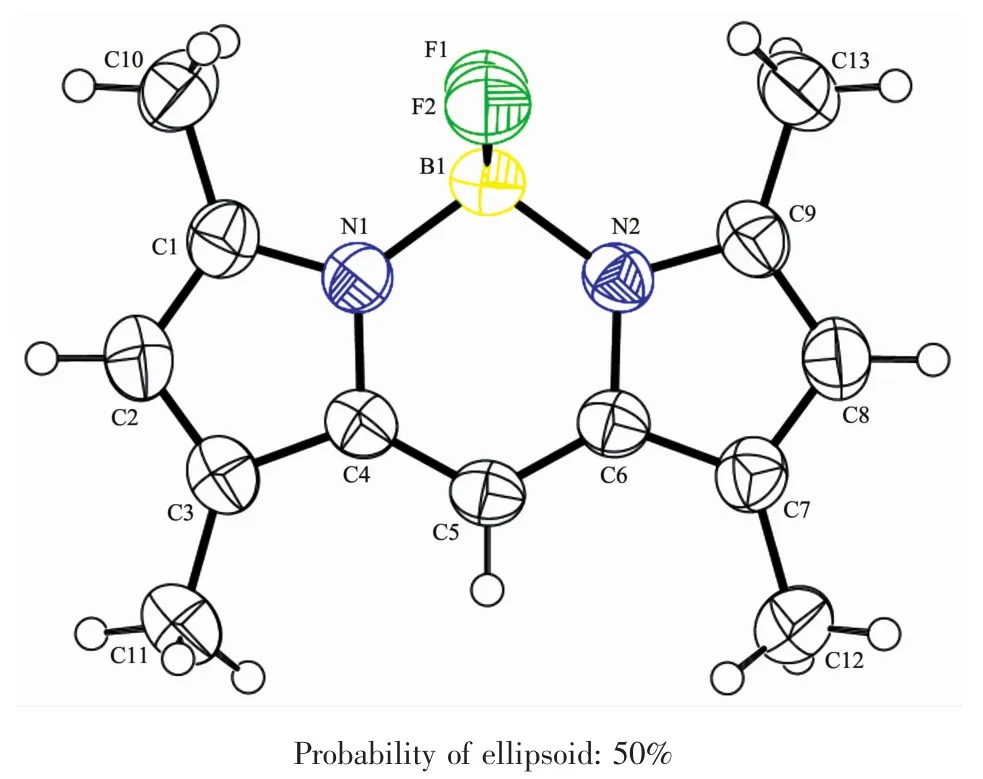
图2 F1的分子结构Fig.2 Molecular structure of F1
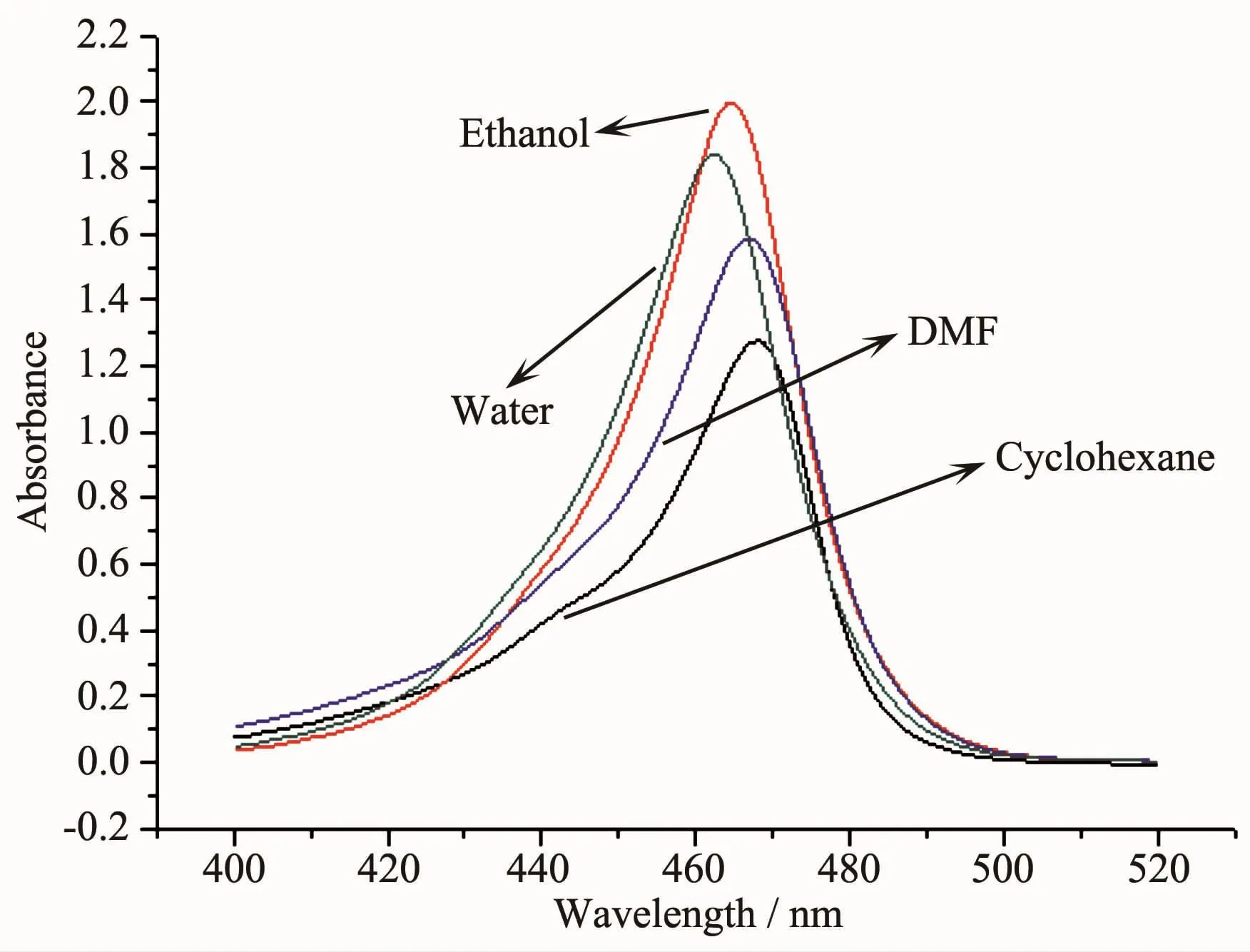
图3 T1在不同溶剂中的紫外可见光谱Fig.3 UV-Vis spectra of T1 in different solvents

图4 F1在不同溶剂中的紫外可见光谱Fig.4 UV-Vis spectra of F1 in different solvents
2.3 荧光光谱
图5为F1的稳态荧光光谱,激发波长为492 nm,其在环己烷、N,N-二甲基甲酰胺、乙醇以及水中的最大发射波长分别为519、516、515和508 nm。从中可以看出,与非极性溶剂环己烷相比,其余3种极性溶剂中F1的发射波长都出现了蓝移,这一原因可以归结为F1是极性分子。对于极性分子F1,在极性溶剂中基态能量降低,第一单重激发态能量也降低但降低幅度小,增加了第一单重激发态与基态之间的能量差,因此其在极性溶剂中更趋于稳定和出现上述蓝移的情况。同时,虽然DMF的极性比乙醇大,但是F1在乙醇中的发射波长更短,与DMF相比出现了蓝移,其可能原因在于乙醇属于质子性溶剂,能和荧光分子中氟原子形成氢键使得基态能量降低较多,增加了能量差。
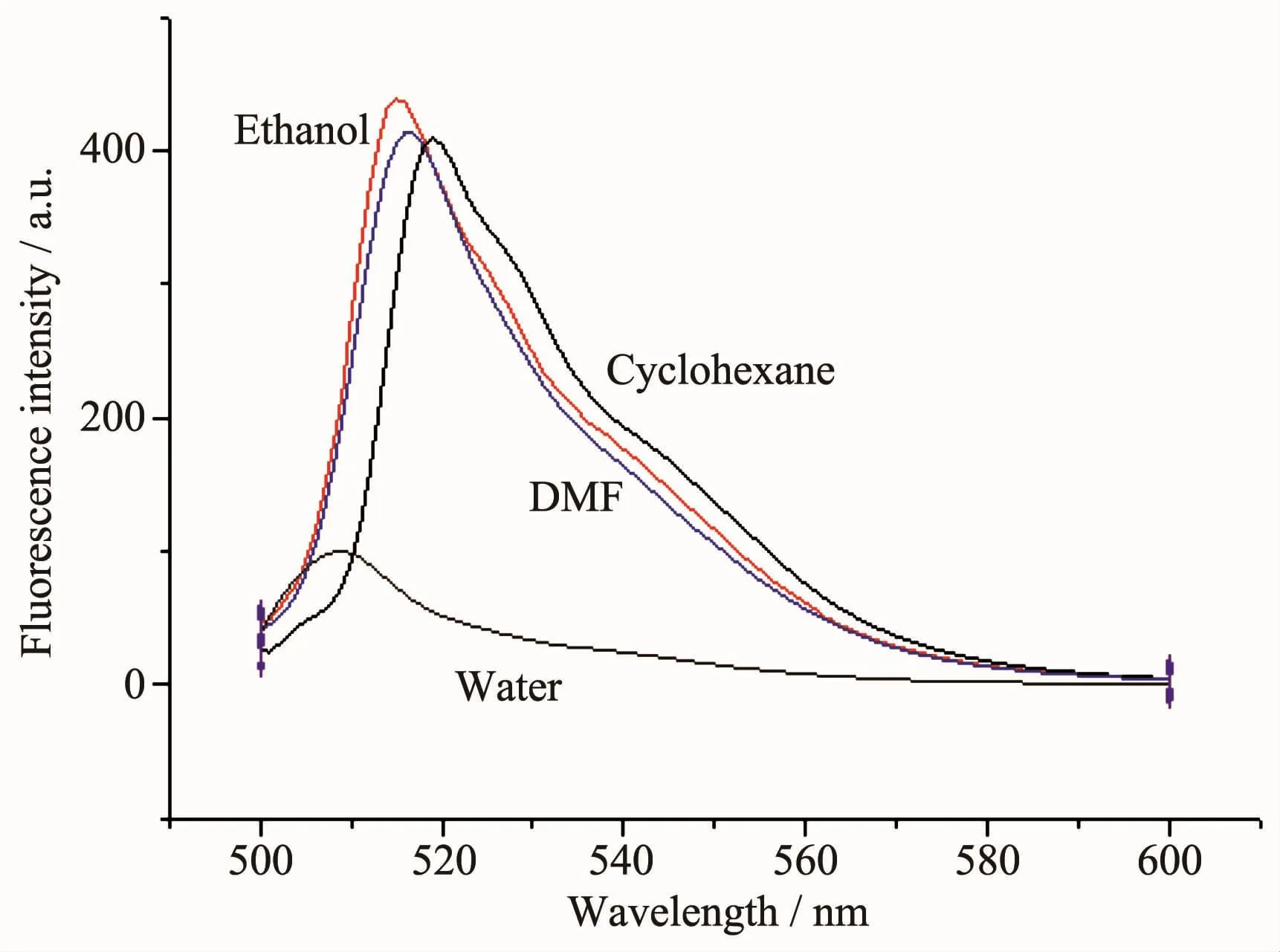
图5 F1在不同溶剂中的荧光光谱Fig.5 Fluorescent spectra of F1 in different solvents
2.4 反应性分析
2.4.1 T1的反应性分析
图6、图7分别为T1在乙醇中的优化构型和分子表面静电势。从图中可以看出T1为离子型化合物(与晶体结构数据相符),有着氟硼化合物所不具有的性质。该化合物相比无对甲苯磺酸保护的二吡咯甲川而言,更加稳定而不易被氧化;其也能在三乙胺存在下,脱除对甲苯磺酸而接上类似氟硼化合物的其他离子结构。
2.4.2 F1的反应性分析
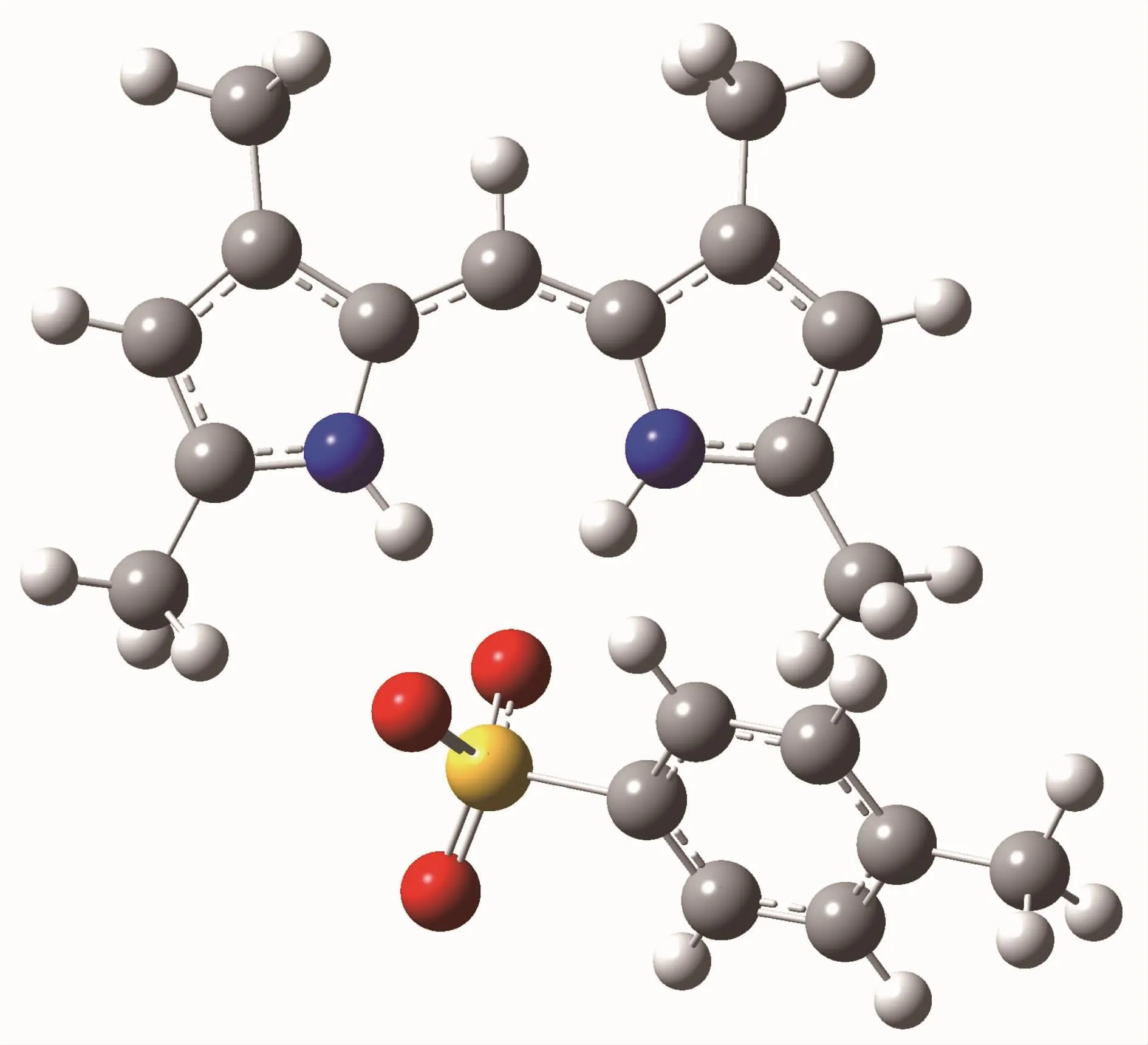
图6 T1在乙醇中的优化构型Fig.6 Optimized configuration of T1 in ethanol
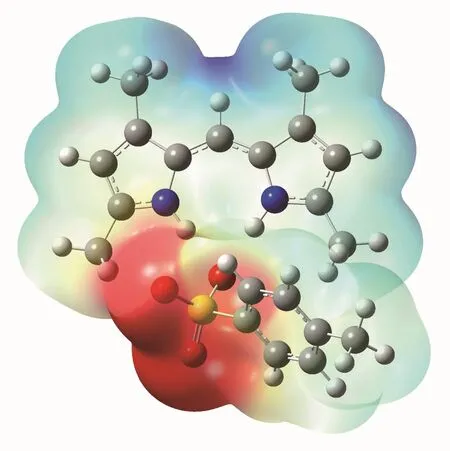
图7 T1在乙醇中的分子表面静电势Fig.7 Molecular surface electrostatic potential of T1 in ethanol
从图8中可以看出,2个吡咯环以及中间的meso位碳处于有效的共轭。优化得出二面角C1-C2-C3-C5:179.897 71°,C1-C7-C8-C9:-179.893 17°,C5-C4-N13-B14:-179.84105°,C9-C10-N12-B14:179.845 11°,并且根据单晶数据可知3个环接近共平面。图9中氟原子周围表面静电势为负,次甲基周围表面静电势为正。虽然基态气相优化中,meso位碳原子的Mulliken电荷[30]为-0.226e,但是其外围的分子表面静电势较正,因此该位置无论是亲电反应还是亲核反应都较难进行。受氟原子影响,亲电试剂优先进攻2,6位碳 (Mulliken电荷为-0.279e),这与Worries和Koek等发现的氟硼二吡咯甲川在2位可引入磺酸基以及2,6位容易在浓硝酸中被硝化相符;另外亲电试剂还可以进攻3,5位甲基,这和Ono和Watanabe将氟硼二吡咯甲川与醛基反应在3位引入双键相符[31]。
从表3中的键长以及上述二面角可以看出,该分子高度对称,对称轴为次甲基的碳原子与硼原子的连线,这一点在核磁位移中也可以看出。与C18和C30相比,C22和C26与吡咯间的碳碳键更接近于单键,这一点可以从晶体结构参数中得到证明,因此吡咯环上的电子离域在前者的程度更大。从表4中的HOMO轨道与LUMO轨道的延展性以及变化来看,亲电试剂优先进攻3,5位甲基以及2,6位碳,而亲核试剂进攻meso位碳。而1,7位甲基上无论是HOMO还是LUMO延展性都很弱而且变化不大,因此当以F1为原料进行反应时,该位置一般不会参与反应。从表3和表4中可以看出,溶剂对键长和前线轨道的影响很小。
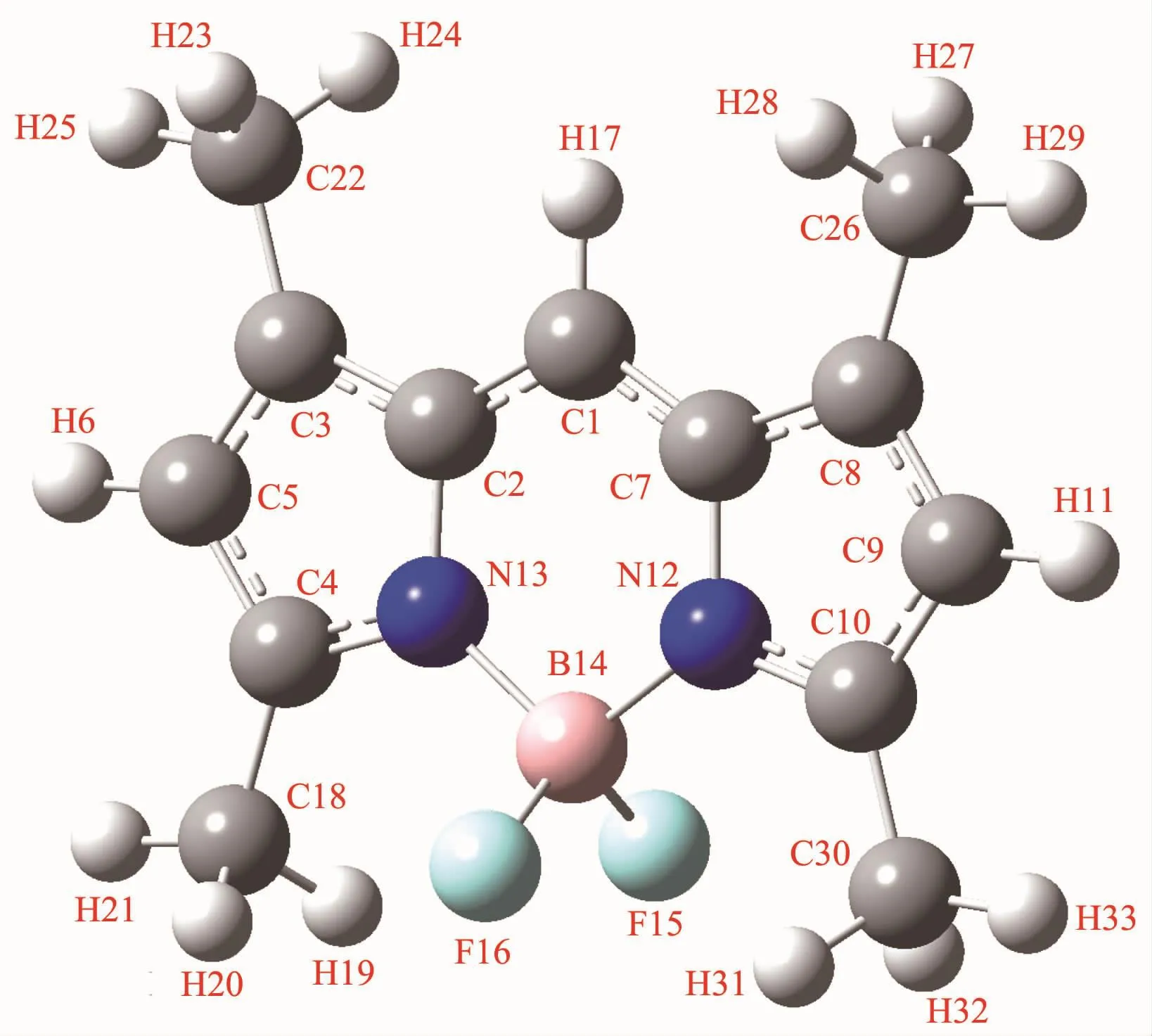
图8 F1在乙醇中的优化构型Fig.8 Optimized configuration of F1 in ethanol
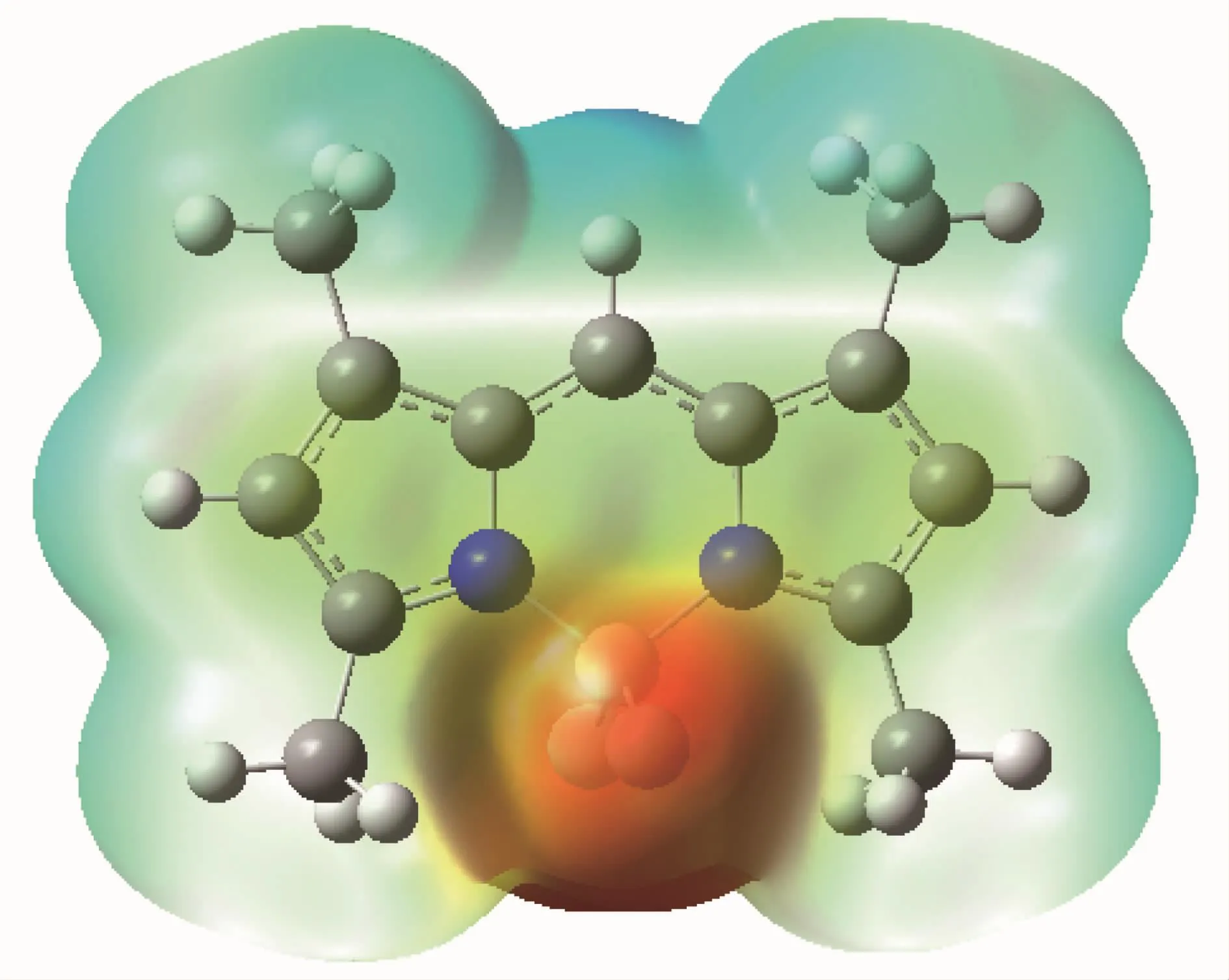
图9 F1在乙醇中的分子表面静电势Fig.9 Molecular surface electrostatic potential of F1 in ethanol

表3 在气相和不同溶剂中F1的部分键长Table 3 Selected bond lengths for F1 in gas phase and different solvents

表4 F1的HOMO轨道和LUMO轨道Table 4 HOMO and LUMO orbitals for F1
3 结 论
本文在合成 1,3,5,7-四甲基对甲苯磺酸二吡咯甲川(T1)和 1,3,5,7-四甲基氟硼二吡咯甲川(F1)的基础上,利用NMR、高分辨质谱和单晶衍射对其进行了表征。晶体衍射结果表明2种化合物的晶体都属于单斜晶系。T1的紫外可见光谱表明,该化合物具有较高的摩尔消光系数。F1的荧光光谱研究表明,其为极性化合物。F1的理论研究表明,该化合物虽然meso位没有取代基但仍具有多种衍生方式,不会因meso位碳上连氢使衍生受到限制。另外,在合成过程中温度要尽量控制在25℃以下,这样产品的产率和纯度都较好。
[1]YU Chang-Jiang(于长江),JIAO Li-Juan(焦莉娟),HAO Er-Hong(郝二红),et al.China Sciencepaper(中国科技论文),2013,8(12):1291-1297
[2]XI Na(袭娜).Thesis for the Master of Nanjing University of Posts and Telecommunications(南京邮电大学硕士学位论文).2012.
[3]KANG Rui-Xue(康瑞雪).Thesis for the Master of Henan University(河南大学硕士论文).2015.
[4]DOU Yuan-Yuan(窦媛媛).Thesis for the Master of Henan University(河南大学硕士论文).2013.
[5]Okada D,Nakamura T,Braam D,et al.ACS Nano,2016,10(7):7058-7063
[6]Chapran M,Angioni E,Findlay N J,et al.ACS Appl.Mater.Interfaces,2017,9(5):4750-4757
[7]Liu K,Zhao X J,Liu Q X,et al.Sens.Actuators B,2017,239:883-889
[8]Yang P,Zhao J Z,Wu W H,et al.J.Org.Chem.,2012,77(14):6166-6178
[9]Gomezduran C F A,Hu R R,Feng G X.et al.ACS Appl.Mater.Interfaces,2015,7(28):15168-15176
[10]Lu J,Ko S B,Walters N R,et al.Org.Lett.,2012,14(22):5660-5663
[11]YI Ping-Gui(易平贵),WANG Tao(王涛),YU Xian-Yong(于贤勇),et al.Chinese J.Inorg.Chem.(无机化学学报),2011,27(3):480-486
[12]Liu G F,Liu J,Sun Z H,et al.Inorg.Chem.,2016,55(16):8025-8030
[13]Poethig A,Strassner T.Organometallics,2012,31(8):3431-3434
[14]HUANG Yan-Ju(黄艳菊),NI Liang(倪良),WANG Qiang(王强),et al.Chinese J.Inorg.Chem.(无机化学学报),2012,28(5):1050-1054
[15]Das S,Hung C H,Goswami S.Inorg.Chem.,2003,42(17):5153-5157
[16]Altenhoff G,Goddard R,Lehmann C W,et al.J.Am.Chem.Soc.,2004,126(46):15195-15201
[17]ZHANG Fu-Xing(张复兴),WANG Jian-Qiu(王剑秋),KUANG Dai-Zhi(邝代治),et al.Chinese J.Inorg.Chem.(无机化学学报),2013,29(3):537-543
[18]Shimizu T,Nakatsu T,Miyairi K,et al.Biochemistry,2002,41(21):6651-6659
[19]Zhou T L,Jia T,Zhao S S,et al.Cryst.Growth Des.,2012,12(1):179-184
[20]Zhu Y Q,Guo Y,Xie D Q.J.Phys.Chem.A,2007,111(38):9387-9392
[21]Zade S S,Bendikov M.Org.Lett.,2006,8(23):5243-5246
[22]Wang S C,Tantillo D J.J.Phys.Chem.A,2007,111(30):7149-7153
[23]Boyd S L,Boyd R J.J.Chem.Theory Comput.,2007,3(1):54-61
[24]Michalkova A,Ilchenko M,Gorb L,et al.J.Phys.Chem.B,2004,108(17):5294-5303
[25]Kovács G,Pápai I.Organometallics,2006,25(4):820-825
[26]Gupta M,Silva E F,Svendsen H F.J.Phys.Chem.B,2012,116(6):1865-1875
[27]Caricato M,Scalmani G,Trucks G W,et al.J.Phys.Chem.Lett.,2010,1(15):2369-2373
[28]Fang S.Ind.Eng.Chem.Res.,2012,51(6):2762-2768
[29]Qu R J,Liu H X,Feng M B,et al.J.Chem.Eng.Data,2012,57(9):2442-2455
[30]XU Guang-Xian(徐光宪),LI Le-Min(黎乐民),WANG De-Min(王德民).Quantum Chemistry:Vol.2.2nd Ed(量子化学:中册.2版).Beijing:Science Press,2009.
[31]Ono M,Watanabe H,Kimura H,et al.ACS Chem.Neurosci.,2012,3(4):319-324
[32]Fukui K,Translated by LI Rong-Sen(李荣森).Chemical Reaction and Electronic Orbit(化学反应与电子轨道).Beijing:Science Press,1985.
[33]Belzile M N,Godin R,Durantini A M,et al.J.Am.Chem.Soc.,2016,138(50):16388-16397
[34]LUO Liang(罗亮),LI Wei(李卫),GAO Ge(高戈),et al.Chem.J.Chinese Universities(高等学校化学学报),2011,32(9):2091-2096
Synthesis,Crystal Structure and Density Functional Theoretical Study of 1,3,5,7-Tetramethyl-4,4-difluoro-4-bora-3a,4a-diaza-s-indacene
DUAN Hai-Chao1YI Ping-Gui*,1YU Xian-Yong1LI Xiao-Fang1LIU Wu1SU Jian2
(1School of Chemistry and Chemical Engineering,Hunan University of Science and Technology,Key Laboratory of Theoretical Organic Chemistry and Functional Molecules of Ministry of Education,Hunan Province College Key Laboratory of QSAR/QSPR,Xiangtan,Hunan 411201,China)
(2School of Chemistry and Chemical Engineering,Nanjing University,Nanjing 210023,China)
1,3,5,7-tetramethyl-3a,4a-diaza-s-indacene p-toluenesulfonate(T1)was synthesized by solvent-free method using 2,4-dimethylpyrrole,triethyl orthoformate and p-toluenesulfonic acid.1,3,5,7-tetramethyl-4,4-difluoro-4-bora-3a,4a-diaza-s-indacene(F1)was synthesized by solvent-free method using T1,boron trifluoride diethyl ether and triethylamine.The structures of compounds have been characterized by1H NMR and single-crystal X-ray diffraction.The results show that T1 belongs to monoclinic system,space group P21/c with a=0.777 3(3)nm,b=1.518 5(5)nm,c=1.612 2(5)nm,β=91.923(12)°,V=1.901 9(11)nm3,Z=4 and that F1 belongs to monoclinic system,space group P21/n with a=0.775 02(1)nm,b=1.444 20(3)nm,c=1.174 35(2)nm,β=107.779 5(9)°,V=1.251 65(4)nm3,Z=4.In four different solvent,the UV-Vis spectrum of T1,the UV-Vis and steady-state fluorescence spectra of F1 were determined.The possible derived way of T1 and F1 were investigated by the density functional calculation and the front orbital theory.CCDC:1578792,T1;1578790,F1.
1,3,5,7-tetramethyl-4,4-difluoro-4-bora-3a,4a-diaza-s-indacene;NMR;crystal structure;fluorescence spectra;density functional theory
O613.8+1
A
1001-4861(2017)12-2201-07
10.11862/CJIC.2017.271
2017-05-15。收修改稿日期:2017-09-11。
国家自然科学基金(No.21172066)、湖南省高校科技创新团队项目(湘教通[2012]318)和湖南省教育厅重点科研项目(No.17A065)资助。
*通信联系人。 E-mail: pgyi@hnust.edu.cn,Tel:+86-731-58290187,Fax:+86-731-58290509;会员登记号:S060006760M。
猜你喜欢
分子催化(2022年1期)2022-11-02
化工设计通讯(2022年5期)2022-05-25
化学工业与工程(2022年1期)2022-03-29
四川轻化工大学学报(自然科学版)(2021年3期)2021-08-30
化学与粘合(2020年4期)2020-09-11
生物工程学报(2020年1期)2020-03-12
国外医药(抗生素分册)(2016年2期)2016-07-12
中国塑料(2016年2期)2016-06-15
中国卫生标准管理(2015年4期)2016-01-14
中国当代医药(2015年36期)2015-03-11
