
高温锂电池氧化物正极材料研究现状与展望
2017-12-13 10:51宋恒旭牛永强赵宇宏
无机化学学报 2017年12期
宋恒旭 牛永强 侯 华 吴 铸 赵宇宏*,
综述
高温锂电池氧化物正极材料研究现状与展望
宋恒旭1牛永强1侯 华1吴 铸2赵宇宏*,1
(1中北大学材料科学与工程学院,高温锂电池材料实验室,太原 030051)
(2中国科学院上海微系统与信息技术研究所,新能源技术中心,上海 200050)
高温锂电池是热电池向中低温度范围的拓展和延伸,在石油、天然气及地热探测等领域有很好的应用前景。相对于具有大比容量和接近纯锂电极电位的锂合金负极材料,正极材料还有不小的发展潜力。因此,正极材料是提升高温锂电池性能的关键材料。而在正极材料中,氧化物材料表现出高电压特性以及高热稳定性,可以推动高温锂电池小型化发展,满足特定条件下的电流电压供给。目前,并没有针对高温锂电池氧化物正极材料的系统性综述。为了促进本领域的快速发展,优化能源结构,本文系统总结了高温锂电池过渡族金属氧化物正极材料的研究进展,包括其物理特性、电化学特性及合成与制备方法,对材料的可利用特性以及不足之处加以说明;进而对氧化物正极材料在高温锂电池领域的应用做出展望。
高温锂电池;正极材料;氧化物;电化学性能;制备
0 引 言
当前全球能源的短缺以及突出的环境问题使能源结构的转变成为社会发展中的重大问题。在一些能源开发的特殊环境中,电池需要耐受高温高压的作用,所以有了对高温锂电池的应用需求。本文所指的高温锂电池是以锂或锂合金为负极,采用低温熔盐作电解质,工作温度比常规锂电池高的一类电池。高温锂电池的工作温度(100~350℃)比军用热电池(350~550℃[1])低,所以同样可以称之为低温热电池。高温锂电池的结构同热电池相似,所以其继承了热电池的一些优良特性,如储存时间长(10~25年)、安全可靠、高比能量、结构紧凑等。不同的是,两者的适用领域不同,热电池的大功率放电和快速激活等特性使之特别适合作为军工领域中的炮弹引信和导弹的电源,并且已经得到广泛的应用与发展[2-3];高温锂电池因为其电解质熔点低,使得电池可以在民用工业领域得到进一步的发展。例如在地热勘探和地下油气开发中一般的水溶液电解质电池和有机溶液电解质电池因为高温而无法正常工作。虽然Li-Mg/SOCl2有机电解质电池(工作温度达到180℃)可以勉强用于石油天然气勘探,但是在更高的温度下已经不适用[4]。而高温锂电池可以很好的解决这一问题。高温锂电池可以利用地热环境中的热量激活和工作,从而可以大幅度简化以热电池为基础的复杂电池结构,提高比容量,同时降低电池生产制造成本。
高温锂电池的发展与电解质体系的发展密不可分。电解质的熔融温度决定了高温锂电池的最低工作温度,电解质体系主要有卤盐和硝酸盐体系。电解质体系要与正负极材料匹配,除此之外高温锂电池的电极材料要满足下面几个条件[5]:① 电极材料有良好的热稳定性,保证在工作温度范围内不会严重氧化和分解;②电极材料尽可能有高的导电率,对导电率低的电极材料能通过添加导电剂改善其导电性能;③电池内部材料相容性好,发生的副反应尽可能的少,使电池发挥出最大的性能。
高温锂电池的正极材料可以借鉴原来的热电池材料,例如早期Ca或Mg系热电池中的正极材料如V2O5和WO3等氧化物,以及在锂离子电池中广泛应用的正极材料,如LiMn2O4等嵌锂化合物。理想的高温锂电池正极材料应具备[6]:
(1)合适的氧化还原电位,与电解质相匹配,防止电解质的氧化。
(2)热稳定性好,在高温工作环境中正极材料不会发生严重的热分解。
(3)电子导电性较好,正极阻抗尽可能的低,或者可以通过添加导电剂来使正极材料的导电性有大的提高。
(4)在电解质中溶解度低,自放电低。
(5)在有水或氧的环境中有较好的稳定性,避免在正极材料表面形成氧化膜。
(6)正极材料的放电产物在电解质中溶解度低。
(7)正极材料能被电解质润湿,以减少彼此接触阻抗。
(8)有尽可能小的密度,以提高电池比容量。
(9)材料廉价易得,成本低。
(10)对环境不会造成污染。
正极材料中的氧化物材料具有高的电势电位,并且一般来说氧化物材料的稳定性较高,而正极材料的热稳定性对高温电池来说是至关重要的。高温锂电池正极材料中的氧化物一般指过渡族金属如Ti、V、Nb、Cr、Mo、W、Mn、Fe、Co、Ni和 Cu 的氧化物。为了获得更稳定的电压和更大的比容量,有时候也对这些氧化物进行锂化处理或掺杂改性。本文将介绍其中应用最广泛和最有前景的几种氧化物。
1 钨的氧化物
1.1 WO3
早期应用在Ca和Mg系热电池中的正极材料是WO3,但是WO3与卤化物电解质不匹配导致其很快就被其他材料替代[1],之后关于WO3的研究很少。WO3在自然界中以钨华或钨赭石矿物态存在。WO3颜色为明亮的黄色,密度为7.21~7.30 g·cm-3,熔点为1 472℃。WO3的晶体结构会随着温度的改变而改变,在-50~17℃为三斜晶系、17~330℃为单斜晶系、330~740℃为正交晶系、740℃以上为四方晶系。单斜晶系的WO3最为常见并且在本文所研究的高温锂电池工作温度范围内,其空间群为P21/n,其晶体结构为八面体(钨原子位于八面体中心位置,氧原子位于八面体顶点位置)。但是这种结构不稳定,在放电的过程中产生畸变会阻碍锂离子的扩散,而且八面体结构的WO3由于扩散截面积小,不利于锂离子的扩散[7]。由钨精矿与氢氧化钠或苏打经过高温熔融或高温压煮制成钨酸钠溶液,再经离子交换或萃取提纯、蒸发工艺制得仲钨酸铵晶体,最后在700℃煅烧制得三氧化钨。仲钨酸铵在氧化剂的作用下分解可用式(1)表示:

WO3的电化学性能将与Li2WO4一同列出,方便对比。
1.2 Li2WO4

图1 四种单体电池化学体系的TGA/DSC热分析曲线:(1)WO3+(LiNO3-KNO3),(2)Li2WO4+(LiNO3-KNO3),(3)WO3+(LiNO3-KNO3-CsNO3),(4)Li2WO4+(LiNO3-KNO3-CsNO3)[8]Fig.1 TG and DSC curves of(1)WO3+binary eutectic,(2)Li2WO4+binary eutectic,(3)WO3+ternary eutectic,(4)Li2WO4+ternary eutectic[8]
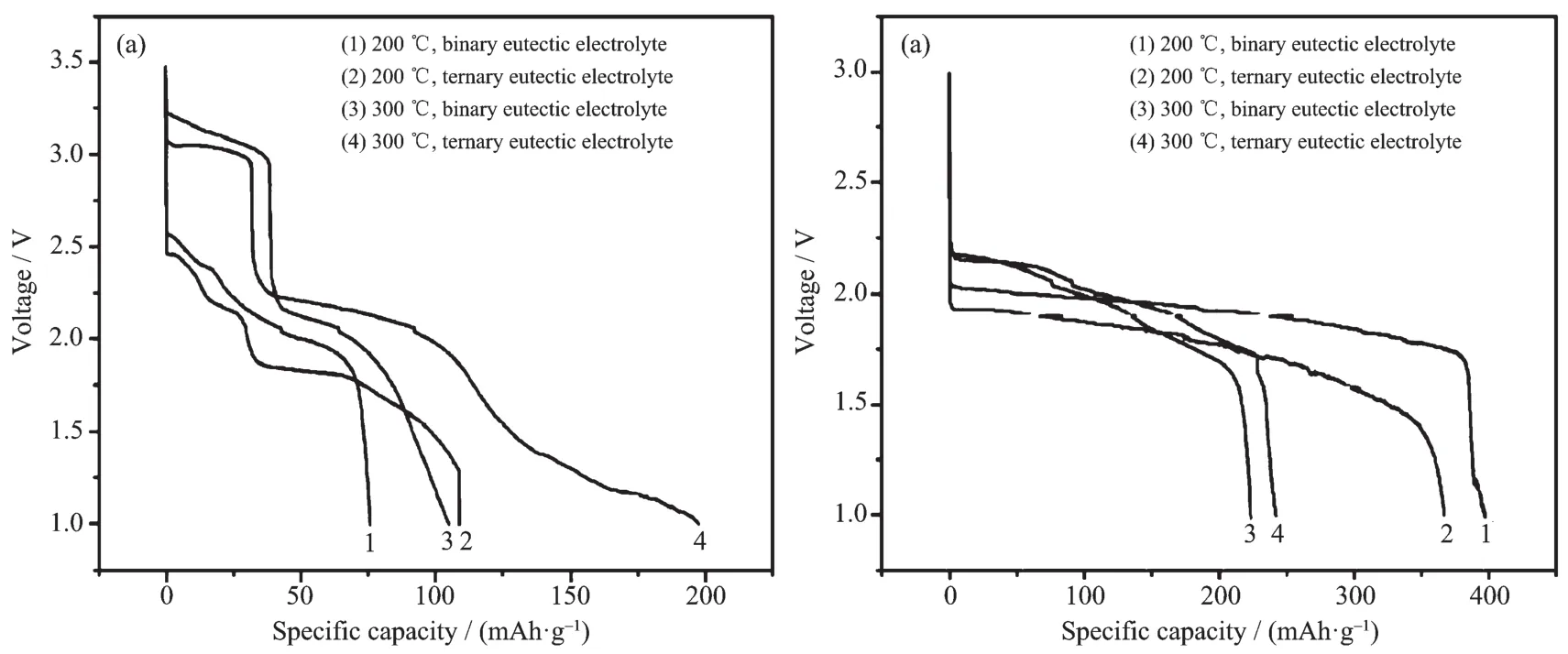
图2 (a)Li-Mg-B/WO3和(b)Li-Mg-B/Li2WO4单体电池恒流放电曲线[8]Fig.2 Galvanostatic discharge profiles of(a)the Li-Mg-B/WO3cells and(b)the Li-Mg-B/Li2WO4cells[8]
Li2WO4是WO3经过锂化处理后得到的材料,属于三角晶系,结构比WO3稳定,锂离子扩散截面也更大。王振杰等[8]分析了WO3和Li2WO4在二元和三元硝酸熔盐中的兼容性,TGA和DSC曲线如图1所示。图1(b)中4种体系中只有WO3+(LiNO3-KNO3)体系在350℃附近出现吸热峰,除此之外只有在电解质熔融温度下出现对应的吸热峰。这表明WO3和Li2WO4正极材料在常用硝酸熔盐中的热稳定温度达到350℃,满足高温锂电池的工作温度范围。文献[8]中还对比了WO3和Li2WO4在低熔点硝酸盐电解质中的放电表现,如图2所示。WO3开始放电电压高,接近3.25 V,但是电压衰减快且电压稳定性差,有多个放电平台,说明WO3正极材料在放电过程中频繁发生相变。经过锂化处理的钨的氧化物初始电压平台较WO3降低0.5 V左右,即锂化处理会降低金属氧化物的电位,但是从图2(b)中可以看到Li2WO4单体电池体系只有一个电压平台,放电电压平稳。当电解质为LiNO3-KNO3,截止电压为1 V时,在200℃下Li2WO4正极材料比容量为397.09 mAh·g-1,这已经接近按式(3)计算得到的理论比容量(408.92 mAh·g-1)。 WO3和 Li2WO4的正极反应[7]分别为式(2)和(3):

Li2WO4的合成方法有软化学法和高温固相法。软化学法反应条件温和,所制备的产物均匀性好,但是适用于软化学法的钨源匮乏,较为简单易行且成熟的还是高温固相法。王振杰[7]采用高温固相法将Li2CO3与WO3的混合物在600℃下保温一定时间,然后在800℃下烧结得到Li2WO4。
2 铬的氧化物
2.1 CaCrO4
高温锂电池中铬的氧化物有CaCrO4、CrO2和Ag2CrO4等。CaCrO4正极材料应用在早期的钙系热电池中。采用LiCl-KCl作电解质,425℃下CaCrO4正极材料相对钙的电极电位为2.65 V。CaCrO4为黄色晶体,随着结晶条件的不同可以产生不同的水合物。无水CaCrO4属于四方晶系,热分解温度在800℃以上,其在水中的溶解度很小,并且随着温度升高溶解度反而降低,不溶于乙醇和丙酮,但在LiCl-KCl熔盐中溶解度较高,在350℃时溶解度为10%,在600℃时溶解度为34%[5],这样高的溶解度会导致溶解在电解质中的CaCrO4与负极反应,从而造成电池严重的自放电[9]。其自放电可用式(4~6)表示[10]:

CaCrO4的放电过程为:CrO42-首先与1个电子作用而被还原为CrO43-,之后CrO43-与Cl-和Ca2+共同作用产生深绿色的五价铬化合物,这一产物会继续发生反应(6),这些产物在电解质中会以树枝状姿态生长,随着反应的进行,树枝状物质长大并可能造成电池的短路[9]。当LiCl-KCl-CaCrO4组分分别为42.8%、52.2%、5%(w/w)时,在 500和 350℃下的电导率分别为1.31和0.59 S·cm-1[11]。CaCrO4在LiCl-KCl中的溶解会导致共熔盐的熔点降低,同时,溶解的CaCrO4又使熔盐的电阻升高。CaCrO4固有的副反应以及在低温时电阻增大使得其在低温领域不再适用,并且铬酸钙有毒,对皮肤和粘液膜都有腐蚀作用。
2.2 CrO2
CrO2是一种半金属铁磁材料,熔点仅为375℃。在250~500℃时分解成三氧化二铬和氧。在250℃下,CrO2在CsBr-LiBr-KBr熔盐中以15.8 mA·cm-2的电流密度放电的电化学性能比锰的氧化物差,在300℃下其放电性能有所改善但是仍然比MnO2和 LiMn2O4差[12]。 CrO2开路电压可达 3.1 V,但是放电时电压下降速率快,实际比容量小。CrO2低的分解温度和差的放电性能使之在高温锂电池中的应用受到限制。
2.3 Ag2CrO4
Ag2CrO4为红棕色单斜结晶或粉末,密度为5.625 g·cm-3,溶于酸、氨水、氰化钾溶液、铬酸碱金属溶液,微溶于水。Ag2CrO4作为高温锂电池的正极材料表现出很大的优势,Ag2CrO4有优良的导电性和热稳定性。Ag2CrO4能够持续承受750 mA·cm-2的电流密度。早在1985年,Mcmanis等[13]就将Ag2CrO4作为正极,研究了其在LiNO3-KNO3熔盐中的放电性能。Mcmanis测试了Ag2CrO4在400℃下电流密度为270~770 mA·cm-2范围内的电化学性能,其放电电压达到3 V,并且在270 mA·cm-2的电流密度下可以在高电位平稳放电达2 min。Guidotti等[14]研究了Li-Si/CsBr-LiBr-KBr/Ag2CrO4电化学体系在250~300℃下电流密度为15.8~32 mA·cm-2范围内的放电性能,结果显示温度对其放电性能影响很大。当电流密度为15.8 mA·cm-2,截止电压为1.25 V时,在250℃下单体电池放电时间为6 min,而在300℃下可以达到10 min以上,如图3所示。然而在接下来的DSC测试中发现Ag2CrO4会与电解质中的溴化物反应放热,所以Ag2CrO4与溴化物电解质并不匹配。Giwa[15]研究了Ag2CrO4在硝酸盐电解质中的放电表现,Ag2CrO4相对Li-Al负极在175℃开路电压可达3.15 V,当截止电压为开路电压的70%时,在215℃时以100 mA·cm-2的电流放电能量可达 110 Wh·kg-1。 随后,Guidotti等[16]又研究了Ag2CrO4在LiNO3-KNO3电解质中的电化学行为,负极分别采用Li-Al和Li-Si合金。DSC测试表明在400℃下Ag2CrO4与LiNO3-KNO3共熔盐不会发生放热反应,说明Ag2CrO4与LiNO3-KNO3共晶熔盐相容性好,满足在特定工作温度范围的需求。负极为Li-Al和Li-Si合金时,在200℃下以7.6 mA·cm-2的放电曲线与电极极化趋势如图4所示,比较发现,负极采用Li-Si合金比Li-Al合金电压平台更高,并且电压平台也更为平稳。在极化方面,以Li-Si合金作负极的体系在第三个放电平台结束后极化才明显上升。采用Li-Si合金作负极,在150~300℃的放电曲线如图5所示,Ag2CrO4作为正极的电极反应可用式(7~9)表示:

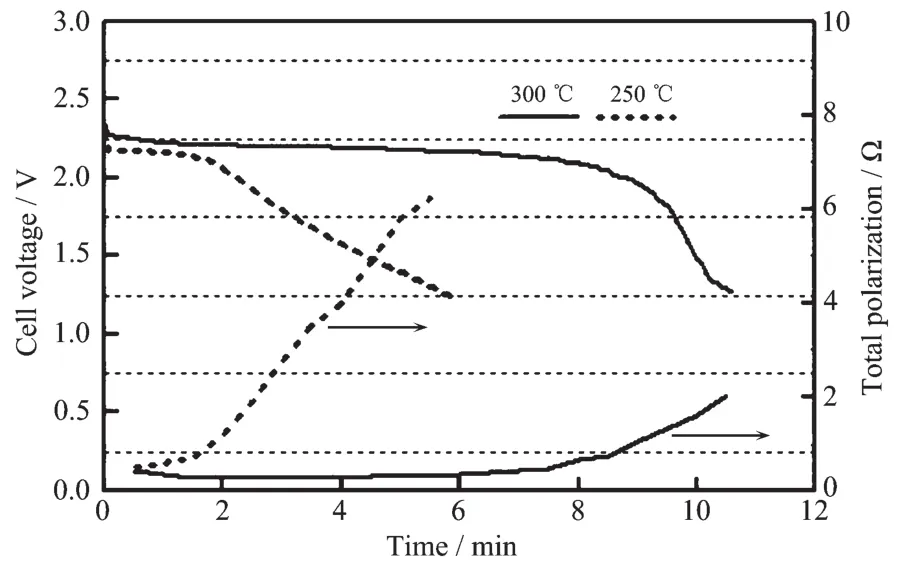
图3 Li-Si/CsBr-LiBr-KBr/Ag2CrO4单体电池在250和300℃以15.8 mA·cm-2的放电曲线[14]Fig.3 Discharge of Li-Si/CsBr-LiBr-KBr/Ag2CrO4single cells at 15.8 mA·cm-2at 250 and 300℃[14]

图4 Li-Al和Li-Si/LiNO3-KNO3/Ag2CrO4体系在LiNO3-KNO3共晶熔盐中以200℃,7.6 mA·cm-2电流密度下的放电表现[16]Fig.4 Comparison of performance of Li(Si)and Li(Al)anodes with Ag2CrO4cathode in molten LiNO3-KNO3eutectic electrolyte at 200℃and 7.6 mA·cm-2[16]

图5 Li-Si/LiNO3-KNO3/Ag2CrO4体系在150~300℃以7.8 mA·cm-2电流密度放电曲线[16]Fig.5 Effect of temperature on the performance of Li-Si/LiNO3-KNO3(MgO)/Ag2CrO4single cells discharged at 7.8 mA·cm-2[16]
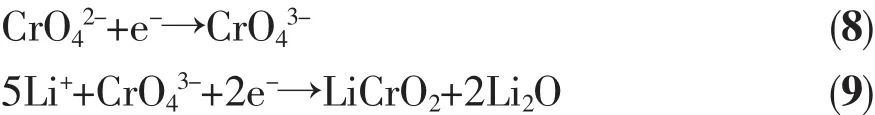
在第一阶段是Ag+的反应,反应如式(7),生成单质银。在第二阶段是Cr6+与1个电子作用变为Cr5+,如式(8)。而在下一步的反应中,Guidotti认为CrO43-与2个电子反应生成 CrO2-,1 mol的 Ag2CrO4与 5 mol的Li+反应。从图5中可以看出Ag2CrO4在200℃的放电性能最佳,第一电压平台在3 V左右,截止电压为1 V时,Ag2CrO4材料的比容量达270 mAh·g-1,并且在200℃还有第四个放电平台,Guidotti认为这很可能是负极的相变所致。
Ag2CrO4常见的制备方法有化学沉淀法[17-18]、水热法[19]、模板法[20]、微波水热法[21]等,在这些制备法中应用最多的是化学沉淀法,因为其工艺步骤相对简单、节能环保、产量大,基本原理是将硝酸银、铬酸钠、氢氧化钠和硝酸原料混合得到沉淀物,然后用去离子水和无水乙醇清洗数次,再将得到的产物真空干燥数小时得到Ag2CrO4[18]。
虽然Ag2CrO4在小电流和大电流放电时都有很好的放电性能,但是Ag2CrO4中Cr6+对环境有污染,而且含有贵金属元素也使得Ag2CrO4正极材料成本高,大规模的应用受到一定限制。
3 钒的氧化物
在过渡族金属中钒有多种价态 (V5+、V4+、V3+、V2+),所以钒可以有多种氧化物,如 V2O5、VO2、V2O3、V6O13等,这些氧化物还可以形成多种嵌锂化合物,而大部分嵌锂化合物为层状结构,有利于Li+的脱嵌[22]。应用在高温锂电池中钒的氧化物包括氧化钒(如 V2O5、VO2等)、 嵌锂化合物 (如 Li1+xV3O8、LiV2O5等)、锂化的氧化钒(LVO)、钒氧碳的复合材料(VOC)和MxVyOz(M为过渡族金属)。本文分别以其中最具代表性的化合物做介绍。
3.1 V2O5
我国的钒资源储量丰富,并且是最大的钒生产国[23]。其中V2O5是应用最广的钒矿产品,V2O5为红色斜方晶体,熔点为658℃,分解温度达到1 750℃,是典型的两性氧化物。V2O5在LiCl-KCl电解质中600℃时溶解度可达17%,且V2O5与LiCl-KCl电解质反应生成氯气和不溶性钒的氧化物,导致电池失效[5]。V2O5在LiCl-KCl电解质中的还原过程如式(10~12):

其电极反应为式(13):

Giwa[15]研究了175~200℃范围内Li-Al/LiNO3-KNO3/V2O5电池体系以35~55 mA·cm-2电流放电的电化学性能。V2O5相对Li-Al的开路电压可达3.5 V,但是在放电时出现明显的电压降,在以35、45、55 mA·cm-2放电的前7 min内平均电压分别为3.0、2.75、2.6 V,放电曲线如图 6 所示。V2O5做正极材料时电压下降很快,所以其比容量较小。另外,V2O5的导电性能差,在作正极材料时一般加入导电剂以增加正极的导电性。V2O5的制备方法有酸法、碱法和盐法等,其中酸法和盐法在生产过程中会产生氯气,对环境造成生态破坏。碱法产生的废气为CO2,对环境影响小,其原理是将钒精矿与纯碱焙烧后生成偏钒酸钠,再与硫酸作用制得V2O5[24]。

图6 不同电流密度下Li-Al/LiNO3-KNO3/V2O5单体电池的放电表现[15]Fig.6 Variation of voltage with current density for Li-Al/LiNO3-KNO3/V2O5cell[15]
3.2 Li1+xV3O8
Li1+xV3O8正极材料是在钒的氧化物中嵌入锂得到的。Li1+xV3O8正极材料具有热稳定性好、导热性好、放电电压高、容量和能量密度高等优点。Li1+xV3O8属于单斜晶系,空间群为P21/m,结构如图7所示,VO6正八面体和畸变的VO5三角双锥通过公用棱边和顶角形成层状的V3O8-结构,再由2个Li1+xV3O8分子组成1个单元晶胞[25]。其热稳定性可达700℃[26]。李志友等[27]将Li1+xV3O8与V2O5对比发现Li1+xV3O8放电的峰值电压虽然有所下降,但是电压平稳性增加,截止电压为峰值电压的75%或2.0 V时的比容量均不低于V2O5。之外,Li1+xV3O8比V2O5的尖峰电压低,电子导电性能更好,Li+扩散容易,阻抗较小。

图7 LiV3O8的结构(010方向)[28]Fig.7 Structure of LiV3O8in projection on to(010)[28]
Li1+xV3O8在放电初期会有放电峰包,即电压先升高后下降的现象。李志友等[29]认为这一现象是因为Li1+xV3O8低的导电性使其在开始放电时由于大的极化而导致电压骤降,反应产生的导电性能好的物质使极化降低,电压再次升高,并且根据XRD的分析结果发现在峰包区有γ-LiV2O5和Li3VO4产物。随着放电的进行,γ-LiV2O5相减少,Li3VO4相增多并且有新相V2O3出现,对此,Li1+xV3O8的放电反应为式(14~15):
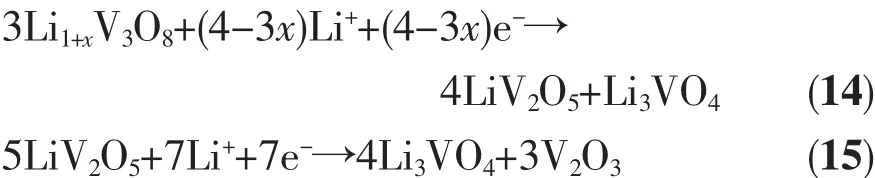
其2个放电阶段(x=0时)对应的理论比容量分别为124.2和198.6 mAh·g-1。反应产物Li3VO4还可以发生反应如式(16):
按照式(16)计算得到Li3VO4的比容量为393.1 mAh·g-1,虽然其理论比容量高,但是因为Li3VO4电子导电率特别低(在500℃时仅为4.34 mS·cm-1[30]),而导致输出电压降至0 V。因此当放电产物完全变为Li3VO4和V2O3时,电池已经因为电压过低而不能被利用。曹笃盟等[31]研究了嵌锂含量不同的Li1+xV3O8材料的高温放电性能,如图8所示,当电流密度较小时电压平稳,但是当电流密度超过400 mA·cm-2时,电压下降较快且电位降低。
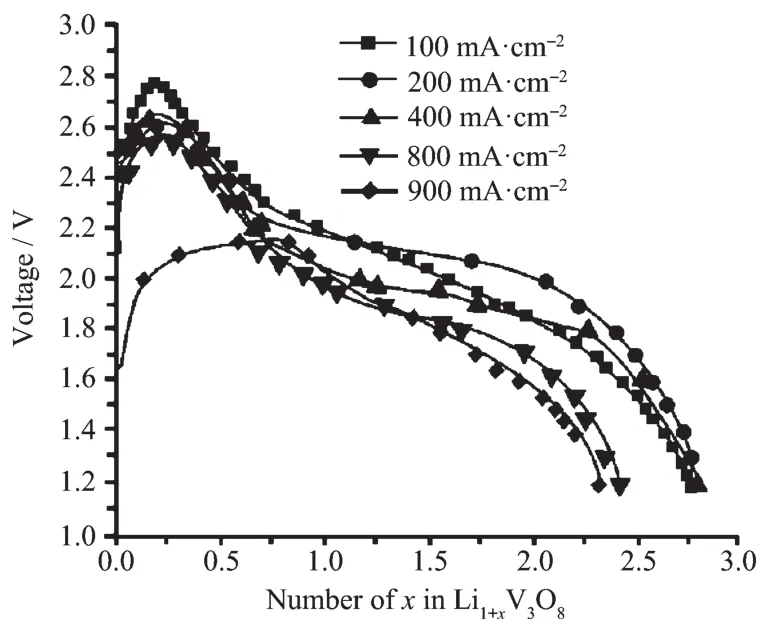
图8 Li1+xV3O8在500℃以不同电流密度放电的放电曲线(截止电压1.2 V)[31]Fig.8 Discharge curves(500℃)of Li1+xV3O8cathode at different current densities(cut-off voltage:1.2 V)[31]
Li1+xV3O8的常规合成方法有高温固相法[28,31-32]、水热法[26,33]、 溶胶凝胶法[22,27,34-35]等。曹笃盟[28]以NH4VO3和Li2CO3为原料采用高温固相法合成LiV3O8,其反应可用式(17)表示:
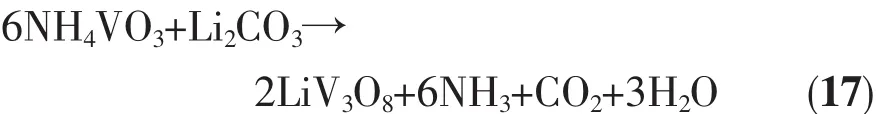
文献[32]中同样利用高温固相合成法,采用Li2CO3与V2O5为材料,按照一定比例混合,其反应温度达680℃,反应时间为24 h。高温固相法的优点是制备流程短,操作简单;缺点是耗能大,所制备的材料均匀性差,粒度不容易控制。文献[26,33]中采用水热反应合成Li1+xV3O8,其制作方法相对简单,Pistoia等[33]将 LiOH与 V2O5在 50℃反应 24~30 h制得Li1+xV3O8粉末。李志友等[27]采用柠檬酸溶胶凝胶工艺制备出了Li1+xV3O8,所得粉末平均尺寸小于20 μm,并且粉末的形态会随着焙烧温度的增加而由蓬松絮状转变为结晶岩状。李海霞等[36]利用V2O5作钒源,将其溶入H2O2溶液中得到V2O5溶胶,再按照比例(nLi∶nV=1∶3)加入 LiOH 溶液,最终通过干燥和烧结制得LiV3O8。液相法与固相法相比过程较为复杂,需要控制的因素多,但是液相法制备出的材料均匀性好,达到分子级别的接触与混合。
锂化的氧化钒(lithiated vanadium oxide,LVO)是LiV2O5和VO2的混合物,在扫描电镜照片[37]中,LVO主要由大量结晶度很高的10 nm左右的片状和块状体组成,其中还杂有非常细小的絮状材料。其特性有[38]:(1)热稳定性好,在700℃以下不会发生分解;(2)电极电位为正,相对锂为2.8 V;(3)不与LiCl-KCl或LiCl-LiBr-KCl等电解质及电解质粘结剂MgO发生不良化学反应。LVO在实际放电过程中发生的反应[37]有:
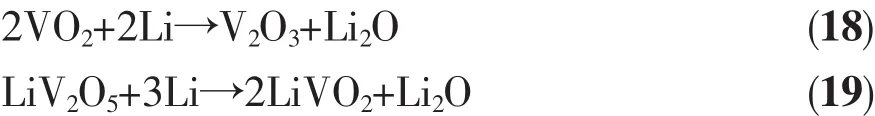
当LVO实际组分为(LiV2O5+4VO2)时,其理论比容量高达 452.2 mAh·g-1[37]。 Gibbard[39]测试了 LVO在LiF-LiCl-LiBr电解质中的电化学性能,在大电流密度下表现良好,8 A·cm-2下的比功率达到20.5 kW·kg-1。刘效疆等[40]研究了在LVO添加一定比例的电解质和二硫化铁后可以在520℃、电流密度为50 mA·cm-2的放电条件下较长时间放电,并研究了LiBr加入量对LVO材料性能的影响。研究表明,LiBr加入量对LVO的晶体结构影响很大,当LiBr实际加入量比理论所需量多20%时,所形成的LVO材料的晶体结构为γ-LiV2O5与VO2的物质,其热稳定性好,分解温度在700℃以上;放电性能良好,当截止电压为1.6 V时放电时间达到61 min。
LVO的制备方法一般是将钒的氧化物与锂化物进行锂化反应制成的。在这里钒的氧化物是指V6O13,为此,先要制备出在V6O13。利用V2O5与V进行反应可以得到V6O13,此反应需要在600℃下加热[38],反应方程为式(20):

在文献[40]中选用偏钒酸铵作为原材料,在惰性气体的保护下,将偏钒酸铵在真空烧结炉里加热至427℃分解制得V6O13材料,反应为式(21):

制得的V6O13再与LiBr在高温和保护气体的条件下发生如式(22)的反应得到锂化的氧化钒。为了保证LiBr与V6O13反应完全,要有足够的保温时间。
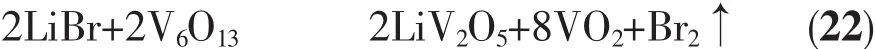
3.4 VOC
VOC(钒氧碳的复合材料)是通过将五氧化二钒和碳复合得到的热电池正极材料,其放电电压平稳且电压较高。其主要活性物质是以LiV2O5和VO2为主的LVO材料的前驱体。在加热和保护气氛的条件下,VOC材料发生的反应[37]有:

在Guibert等[41]的研究中VOC/Li在450℃,100 mA·cm-2的放电条件下可以以2.45 V电压稳定放电。VOC与V2O5相比,放电电压有所降低,但是电压平稳性得到很大改善。Guibert等[41]将V2O5与质量分数分别为5%~10%的石墨粉和30%~50%的LiCl-KCl电解质在365~650℃下通氩气加热15~120 min制得VOC,热分析结果表明VOC比V2O5材料有更好的热稳定性。石奎等[42]对VOC进行形貌分析发现粉末颗粒径向粒径小、比表面积较大的层片状及细棒状材料可以使Li+快速扩散,使电池能够在较大电流密度下以较高的电压放电。
总而言之,把数学建模的思想方法融入到高职数学教学中是创新时代对人才培养的要求,是社会发展的必然结果,这是必要的,也是可行的。通过实践,数学建模思想的应用更有利于学生学习和掌握高职数学的基本知识,激发学生对数学的学习兴趣,而且进一步培养了学生的创新意识和创新能力。随着数学建模对高职数学教学的意义逐渐深入研究,可以看出数学建模思想在提高职高的学生数学素质起到了一定的推动作用。
3.5 钒酸铜
上面列出的几种的钒的氧化物电压比传统热电池中的二硫化铁正极材料电压高,近来对于钒酸铜的研究进一步提高了电池的电压。钒酸铜是通过将CuO与V2O5按照一定比例混合并在一定条件下制 得 的 , 主 要 包 括 CuV2O6、Cu2V2O7、Cu3V2O8和Cu5V2O10等。CuO与V2O5的相图如图9所示。钒酸铜正极材料将CuO所具有的大容量(1 847 mAh·g-1[44])与V2O5所具有的高电压(OCV达3.5 V[15])特性结合起来,表现出良好的电化学性能。另外,钒酸铜材料有很好的热稳定性,CuV2O6和Cu2V2O7的热稳定性都超过600℃[43]。

图9 CuO-V2O5的相图[43]Fig.9 CuO-V2O5phase diagram[43]
Tomer等[43]研究了 α-CuV2O6、α-Cu2V2O7、Cu3V2O8和Cu5V2O10材料在室温下以0.1 mA·cm-2放电(电解质为EC/DMC)和525℃下以100和310 mA·g-1放电的电化学性能。室温下4种材料以0.1 mA·cm-2的放电曲线如图10所示,其中α-Cu2V2O7的比容量最大, 达到 400 mAh·g-1。 高温下 α-CuV2O6和 α-Cu2V2O7的放电曲线分别如图11(a)、(b)所示,在100 mA·g-1的电流密度下2种材料的电压平台都在3.2 V左右,而在310 mA·g-1的电流密度下两种材料的电压平台为2.5 V左右,可见电流密度对实际放电电压影响较大。另外2种材料同在100 mA·g-1的电流密度下放电,α-CuV2O6的电压平台更长,说明以小电流放电时,α-CuV2O6更具优势。Dai等[45]研究了薄 膜 Cu3V2O8和 Cu5V2O10在 LiPO3·Li2SO4·Li2CO3·LiF四元电解质中的放电行为,但同样是在500℃的高温环境下进行测试,Cu5V2O10的开路电压达到3.16 V,并且在1 V的截止电压下比容量为450 mAh·g-1。 随后,Dai等[46]还将 Cu3V2O8制成成品热电池,对其进行恒流(电流密度为340 mA·cm-2)与脉冲(电流密度为990 mA·cm-2)放电测试,成品电池的电压仍然可以达到较高水平,但是电池寿命短,只有2 min左右。如何使电池保持平稳的电压和延长其寿命还需要大量的研究与工作。近来,国内关于钒酸铜材料做高温锂电池正极的研究增多。2014年,闫妍[47]和骆柬氽[48]分别研究了α-CuV2O6和Cu3V2O8在LiPO3-Li2SO4-Li2CO3-LiF电解质中的电化学行为。当放电电流为20 mA·cm-2时,α-CuV2O6单体电池放电电压峰值为2.79 V,电压平台为2.55~2.79 V[47]。骆柬氽[48]测试了 Cu3V2O8在 50、100 和 200 mA·cm-2恒流放电下的单体电压和比容量,其中电流密度为50 mA·cm-2时单体电压为2.9 V,比容量为550 mAh·g-1。 之后,丁晓儒[49]和袁朝军[50-51]研究了 Cu3V2O8在硝酸共熔盐电解质中(温度为190~280℃)的电化学性能,他们的研究都表明Cu3V2O8在190~220℃的范围内电压下降快,无可利用的电压平台,在250~280℃电压比较平稳。袁朝军[52]研究了电解质添加量和导电剂对正极材料放电性能的影响,发现电解质适宜添加量为30%,而活性炭、乙炔黑、CNTs等碳材料都可以明显改善正极材料的放电性能,其适宜添加量分别为4%、3%、3%。

图 10 α-CuV2O6、α-Cu2V2O7、Cu3V2O8和 Cu5V2O10材料在室温下以0.1 mA·cm-2电流密度恒流放电曲线[43]Fig.10 Galvanostatic discharge curves of α-CuV2O6,α-Cu2V2O7,Cu3V2O8and Cu5V2O10at room temperature,at a current density of 0.1 mA·cm-2[43]

图11 α-CuV2O6(a)和α-Cu2V2O7(b)在525℃下以100和310 mA·g-1的电流密度恒流放电曲线[43]Fig.11 High temperature galvanostatic discharge curves of α-CuV2O6(a)and α-Cu2V2O7(b)performed at current densities of 100 and 310 mA·g-1[43]

图12 (a,c)为α-CuV2O6的SEM图像;(b,d)为α-CuV2O6单个颗粒的SEM分层图像;(e)为α-Cu5V2O10颗粒状SEM图;(f)代表单个α-Cu5V2O10颗粒层状图像[43]Fig.12 SEM images of α-CuV2O6particles(a,c);a single α-CuV2O6particle representing the layered structure characteristics(b,d);α-Cu5V2O10particles(e);a single α-Cu5V2O10particle,representing the layered structure characteristics(f)[43]
在钒酸铜的制备上有固相反应法[43,45,47-52]、水热合成法[53]、溶胶凝胶法[54-55]、化学共沉淀法[56]等多种方法,但是在高温锂电池中目前多采用高温固相法[43,45,47-52]将CuO与V2O5按照一定的比例混合均匀并在高温下焙烧得到。Tomer等[43]利用高温固相法将物质的量之比为 1∶1、2∶1、3∶1 和 5∶1 的 CuO 与V2O5混合, 分别合成了 CuV2O6、Cu2V2O7、Cu3V2O8和Cu5V2O10,合成的材料结晶度良好,其中CuV2O6与Cu5V2O10的SEM图像如图12所示。孙榕等[56]利用沉淀法将 Cu(NO3)2·3H2O和NH4VO3按照物质的量之比3∶2配制,通过改变溶液的pH值得到不同的沉淀物,然后通过洗涤、过滤、干燥等步骤,最后在马弗炉中600~700℃焙烧得到片状的Cu3V2O8材料。文献[54]中采用溶胶凝胶法合成Cu3V2O8,基本过程为:将(CuCO3·Cu(OH)2·xH2O)、NH4VO3和柠檬酸按物质的量之比1.5∶2∶1.5混合,把混合物置于80℃下磁力搅拌;将搅拌均匀的悬浊液放入烘干箱蒸干水分得到湿凝胶,将湿凝胶真空干燥12 h获得前驱体,再在马弗炉中650℃下加热10 h,最后冷却研磨得到Cu3V2O8材料。由此获得的Cu3V2O8材料为不规则的块状,粒径尺寸在5~15 μm左右,分布相对均匀。通过不同制备方法可以获得不同形貌的钒酸铜材料,但是不同形貌的材料在高温锂电池中的电化学性能还有待进一步的实验研究。
4 锰的氧化物
用于高温锂电池的锰的氧化物主要是MnO2和LiMn2O4。作为正极材料,锰的氧化物价格低廉而且对环境相对友好,有很好的应用前景。
4.1 MnO2
MnO2是两性氧化物,在常温下为稳定的黑色粉末状物质,无熔点,在535℃发生分解。其晶体结构为八面体结构,O位于八面体的顶角位置,Mn位于八面体中心位置。八面体间通过共棱形成单链或双链,而链间通过共用顶点形成空隙的隧道结构。MnO2有α、β、γ等多种晶体结构,其结构会随着温度的改变而发生转变,例如γ-MnO2在300℃开始向β-MnO2转变,在350~450℃经过适当时间的保温会全部转变成β-MnO2[57]。在高温锂电池中应用的MnO2要通过热处理去除结晶水,并且其结构应在高温的工作环境下保持稳定,这样β型结构最为合适。MnO2作为正极材料电极电位正,在LiCl-KCl电解质中相对Li-Si合金开路电压为3.24 V,初始放电电压为 3.09 V[44],而在 LiNO3-KNO3-KNO2-Ca(NO3)2电解质中相对Li-Mg-B合金开路电压可达3.40 V[58]。Guidotti等[12]研究了MnO2在CsBr-LiBr-KBr电解质中的放电特性,以恒电流15.8 mA·cm-2放电时,在250 ℃下其比容量为 27.8 mAh·g-1(100 C·g-1),而在300 ℃下的比容量增加到 88.9 mAh·g-1(320 C·g-1),截止电压为2 V。随后Guidotti等[59]在以上体系中比较了不同温度和电流密度下MnO2的电化学性能,如图13所示,截止电压为1 V,在300℃和16 mA·cm-2下放电比容量最大为 222.2 mAh·g-1(800 C·g-1),而极化也是最小。在熔点更低的硝酸熔盐中,MnO2是与之匹配的正极材料,在室温至400℃的DSC曲线上MnO2未显示出与电解质反应而产生放热现象[59]。Guidotti等[60]测试了 Li(Al)/LiNO3-KNO3/MnO2体系在温度为150~300℃,电流密度为7.6~30.4 mA·cm-2范围内的放电特性,结果表明,温度对电池体系的放电性能影响很大,在200℃下电池放电性能最佳;电流密度较小时正极材料的比容量更大,当电流密度为7.6 mA·cm-2时比容量达到 144.4 mAh·g-1(520 C·g-1)。本课题组的Niu等[61]研究了以Li-Mg-B作负极,LiNO3-KNO3作电解质的高温锂电池中MnO2的性能表现,其测试温度范围为150~300℃,电流密度为10~30 mA·cm-2。不同温度下以10 mA·cm-2恒流放电曲线见图14,图中MnO2有3个明显的放电平台分别为2.90、2.70和2.10 V。3个放电平台对应的正极反应分别为式(27~29)[62]:

图13 温度和电流密度对Li(Si)/CsBr-LiBr-KBr/MnO2体系放电性能的影响[59]Fig.13 Effect of temperature and current density on the discharge of Li(Si)/CsBr-LiBr-KBr/MnO2cells[59]

图14 不同温度下Li-Mg-B/LiNO3-KNO3/MnO2单体电池以10 mA·cm-2恒流放电曲线[61]Fig.14 Effect of temperature on the performance of Li-Mg-B alloy/LiNO3-KNO3/MnO2single cells discharged at 10 mA·cm-2[61]

第4个很短的平台反应为式(30)[62]:

单体电池在200和250℃下可以长时间高电压放电,但是在150和300℃下表现不佳,主要是因为低温下电解质离子导电率很低,而高温下电池的自放电严重,导致电压和容量大幅度下降。按照式(27~29)计算得到MnO2的理论比容量为924.9 mAh·g-1,而根据实际放电测试得到的MnO2的比容量可以达到 1 052.0 mAh·g-1,Niu 等[61]认为这是由于部分Li2MnO3转化成Li2+yMn2O4,从而导致实际比容量有所增加。另外值得注意的是MnO2材料电压平台相对较多,说明在电池工作过程中正极发生多次相变,放电产物导致极化增大。
MnO2主要取自天然矿物软锰矿,一般采用高温硫酸锰溶液电解法制取,碳酸锰矿和软锰矿均可作为原料。硫酸锰溶液的制备包括浸取、除铁、中和、除重金属、过滤、静置除钙镁等工序,经高温电解后制得粗产品,再经处理包括剥离、粉碎、洗涤、中和与干燥等过程制得合格晶。此外,也有人采用软化学法[63-64]制备出纳米级别的MnO2来提高电池性能,但是在高温锂电池中还有待于研究和应用。
4.2 LiMn2O4
为了得到更为平稳的电压,也有将锰的氧化物锂化处理后作为正极材料。最初应用在锂离子电池中的LiMn2O4在高温锂电池中也有不俗的表现。LiMn2O4有层状结构和尖晶石结构。其中尖晶石结构的LiMn2O4更加稳定(热分解温度达到700℃),属于立方晶系,具有三维隧道结构,更容易实现Li+在其中的脱嵌。尖晶石结构的LiMn2O4与MnO2相比,其与低熔点的硝酸盐相容性更高,达到450℃左右。Guidotti等[12]研究了 LiMn2O4在 CsBr-LiBr-KBr电解质中的电化学性能,300℃下LiMn2O4有比MnO2更平稳的电压和更大的放电容量。在文献[59]中,MnO2和LiMn2O4放电曲线和极化曲线如图15所示,可以看出LiMn2O4电压稳定在2.4 V左右,极化只是MnO2的一半。LiMn2O4在硝酸熔盐中的开路电压在2.9 V左右[7],比 MnO2低 0.5 V左右,在200、250和300℃的工作温度下放电性能最佳的是250℃,截止电压为1 V时比容量达到459.57 mAh·g-1。王振杰[7]推测出LiMn2O4分步放电过程为式(31~33):

而总反应过程可用式(34)表示:
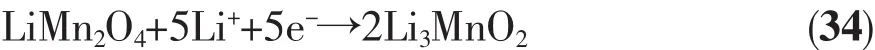
按照式(34)计算得到LiMn2O4材料的理论比容量为 739.86 mAh·g-1。
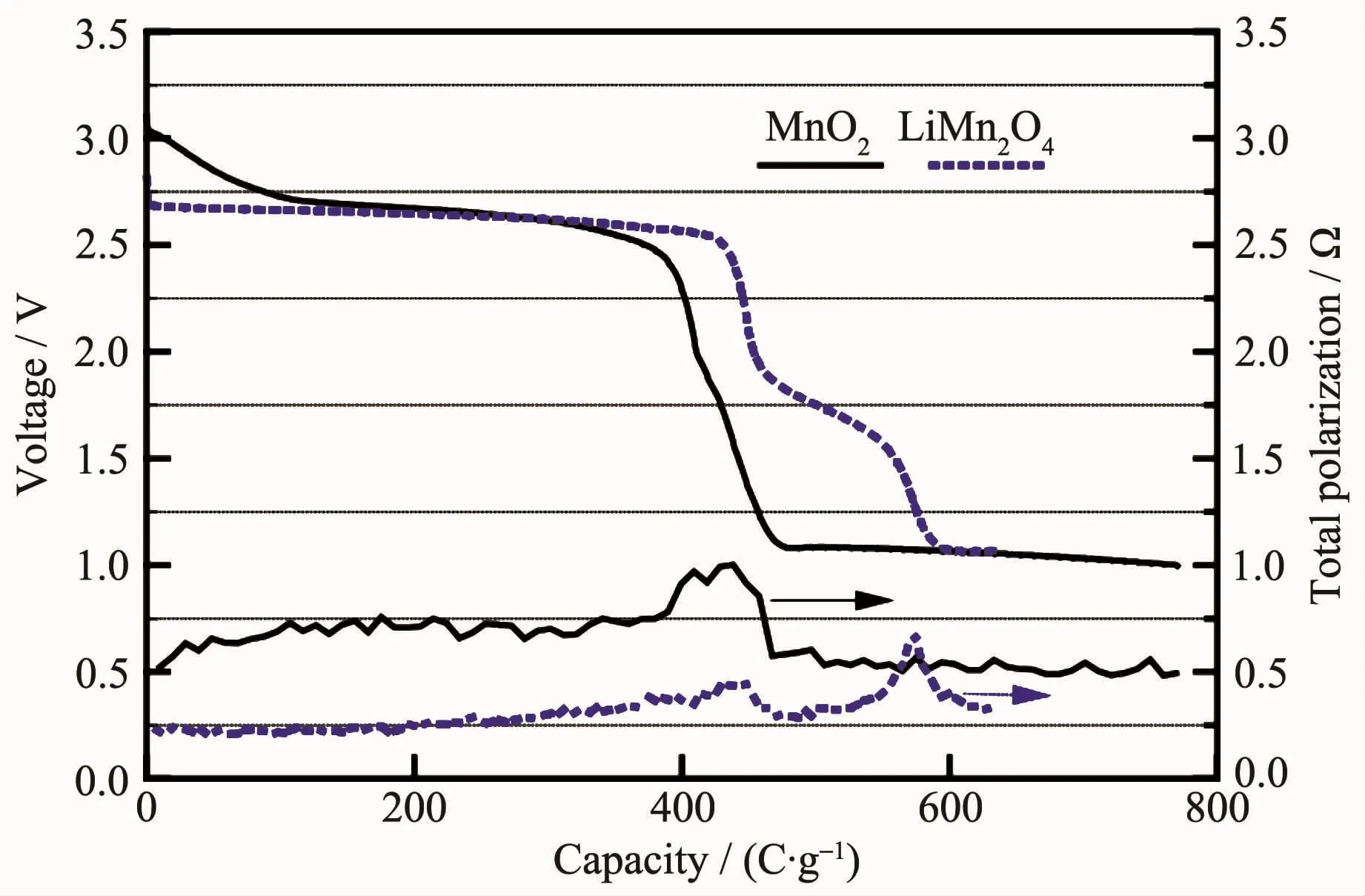
图 15 在 300℃和 16 mA·cm-2下,Li(Si)/CsBr-LiBr-KBr/MnO2和Li(Si)/CsBr-LiBr-KBr/LiMn2O4的放电曲线和极化曲线[59]Fig.15 Relative performance of Li(Si)/CsBr-LiBr-KBr/MnO2and Li(Si)/CsBr-LiBr-KBr/LiMn2O4cells at 300℃ and 16 mA·cm-2[59]
在高温锂电池中所用的LiMn2O4一般是尖晶石结构,其合成方法主要有高温固相法[65-68]、水热法[69]和溶胶凝胶合成法[70]等。其中高温固相法为常用的简单易行的方法,固相合成LiMn2O4的锰源一般是Mn3O4或者MnO2。白亮飞[66]通过将MnO2和Li2CO3按照一定比例混合并研磨2 h,然后将得到的粉末材料压片并在高温下2次退火制得LiMn2O4。Wang等[70]采用溶胶凝胶法将醋酸盐与柠檬酸混合得到凝胶,再将凝胶干燥和焙烧制备出LiMn2O4。Wang等[70]还按此方法用Co掺杂LiMn2O4使得正极材料的实际放电容量有大的提升。高温固相法的缺点是合成材料的均匀性差,而水热法和溶胶凝胶法制备的材料均匀性好,粒度小,但是可控性较差。
5 展 望
高温锂电池应用主要面向石油天然气等地下资源的开发利用、矿山钻井设备的小电流供电,还有油气管道检测、地震和海洋的检测与测量、民用上的轮胎检测和地下测压等领域。目前的常规锂电池工作温度(-55~70℃)太低,而军用热电池工作温度(350~550℃)过高,在此温度和环境下都无法工作,高温锂电池的开发应用就是要满足在这一温度范围内的需求。这也就对高温锂电池正极材料的特点提出了要求,主要是:
(1)高电压。传统的低电压电池会因为堆叠更多的单体电池而使得电池体积较大,高电压的正极输出可以通过减少单体电池层数满足对特定电压需求。这样可以尽可能减小电池体积,同时因为单体电池堆叠较少使得电池的安全性能提高。在高电位的正极材料中,氧化物的电位较高,并且稳定性好,尤其是Mn和V的氧化物及其一系列改性材料,在小电流放电中可以实现单体电池的3 V左右的电压输出,相比传统FeS2(1.8 V左右)的电压平台,其电压高出60%~70%。
(2)大容量。避免频繁更换电池可以提高工作效率并且降低成本。一些氧化物虽然有高的电位和理论比容量,但实际比容量与理论比容量差距较大,对此可以采用掺杂相应的离子或分子集团使材料的晶体结构发生有利改变,或者通过复合材料来使几种相协同放电,比如氧化钒与锂化的氧化钒复合得到的LVO材料要比单相材料发挥出更好的性能,同时具有高电压与较大比容量。
(3)制备简单、环保。材料的来源广泛与制备工艺的成熟使正极材料可以得到更为广泛的应用,有利于商业化发展。环保型材料更是当今的发展趋势。V与Mn的氧化物有相对较广泛的材料来源,并且对环境比较友好,在材料的制备上通过优化工艺降低成本,使其实现工业化大批量生产,将对电池和能源发展起到强有力的推动作用。
(4)对环境要求低。正极材料在制备过程中对环境的要求尽可能的低,例如可以在空气中制备,而不是在惰性气体的保护下制备,这有利于降低生产的成本和实际操作过程的繁杂程度,在大规模生产上也有切实可行性。
综观上述氧化物材料,WO3正极材料的电压平台短并且电压衰减过快,没有实际利用价值。但是经过锂化处理后得到的Li2WO4材料的电压平台稳定、容量大幅度提升,加之原料价格便宜,利用价值较大。铬的氧化物中目前表现最好的是Ag2CrO4材料,其电化学性能好,但是成本较高、环境污染大,不是电池发展的主流方向。钒与锰的氧化物电极电位正,高温下稳定性好,但是基本材料如V2O5和MnO2在放电过程中都存在电压不稳定的问题,经过改进处理后所得到的材料性能有大幅度提升。目前在容量和电压方面表现都较好的是钒酸铜和LiMn2O4,其性能的改进有待进一步的研究和测试。应该说明的是氧化物正极普遍存在导电性差的问题,通常采用的办法是通过掺杂导电剂如乙炔黑、石墨粉等来提高正极的电子导电性或者加入电解质提高离子导电性和降低阻抗。此外,许多氧化物实际放电容量与理论容量相比还有较大差距,其放电机制还需开展深入研究。
[1]Guidotti R A,Masset P.J.Power Sources,2006,161(2):1443-1449
[2]ZHAO Guang-Bo(赵广波),XU Ying(徐莹),BIAN Ji-Qing(汴纪庆),et al.Journal of Detectionamp;Control(探测与控制学报),2008,30(6):64-68
[3]ZHANG Chun-Xiao(张春晓),MU Shi-Bo(穆仕博).Chin.J.Power Sources(电源技术),2010,34(6):614-615
[4]Guidotti R A.35th Intersociety Energy Conversion Engineering Conference and Exhibit:Vol.2.New York:IEEE,2000:1276-1286
[5]LU Rui-Sheng(陆瑞生),LIU Xiao-Jiang(刘效疆).Thermal Batteries(热电池).Beijing:National Defense Industry Press,2005.
[6]Masset P J,Guidotti R A.J.Power Sources,2008,177(2):595-609
[7]WANG Zhen-Jie(王振杰).Thesis for the Doctorate of University of Chinese Academy of Sciences(中国科学院大学博士论文).2014.
[8]Wang Z J,Du J L,Li Z L,et al.Asian J.Chem.,2014,26(23):7939-7942
[9]Masset P J,Guidotti R A.J.Power Sources,2008,178(1):456-466
[10]Guidotti R A,Reinhardt F W,Tallant D R,et al.Evolution,1984,62(3):531-548
[11]Clark R P,Goldsmith H J,Blucher R L.J.Chem.Eng.Data,2002,15(2):277-280
[12]Guidotti R A,Reinhardt F W.38th Power Sources Conference.Albuquerque:Sandia National Laboratories,2000:211-226
[13]Mcmanis G E,Miles M H,Fletcher A N.J.Power Sources,1985,16(4):243-251
[14]Guidotti R A,Reinhardt F W.Proceedings of the International Symposium.Trulove P.C Ed.,Albuquerque:Sandia National Laboratories,1999:451-460
[15]Giwa C O.Materials Science Forum:Vols.73-75.Chemla M,Devilliers D Ed.,Zurich:Trans Tech Publication Ltd.,1991:699-706
[16]Guidotti R A,Reinhardt F W.201st Electrochemical Society Meeting.Albuquerque:Sandia National Laboratories,2002.[17]Soofivand F,Mohandes F,Salavati-Niasari M.Micro Nano Lett.,2012,7(3):283-286
[18]ZHOU Wen-Min(周文敏),HUANG Jian-Feng(黄剑锋),LI Jia-Yin(李嘉胤),et al.J.Synth.Cryst.(人工晶体学报),2015,44(4):954-960
[19]Xu D,Cao S,Zhang J,et al.Beilstein J.Nanotechnol.,2014,5(5):658-666
[20]Liu J K,Luo C X,Quan N J.J.Nanopart.Res.,2007,10(3):531-535
[21]Liu Y,Yu H,Cai M,et al.Catal.Commun.,2012,26(35):63-67
[22]ZHANG Meng-Xiong(张孟雄),ZHANG You-Xiang(张友祥).Chinese J.Inorg.Chem.(无机化学学报),2012,28(10):2065-2070
[23]XIA Yu-Dan(夏玉丹),SONG Hui(宋辉),YANG Bin(杨斌).Guizhou Chemical Industry(贵州化工),2011,36(4):17-18
[24]ZHANG Yun-Hua(张蕴华).Guangdong Chemical Industry(广东化工),2006,33(4):77-79
[25]Wadsley A D.Acta Crystallogr.,1957,10(4):261-267
[26]Dai J,Li S F Y,Gao Z,et al.J.Electrochem.Soc.,1998,145(9):3057-3062
[27]LI Zhi-You(李志友),HUANG Bo-Yun(黄伯云),TANG Chun-Feng(汤春峰),et al.Materials Science and Engineering of Powder Metallurgy(粉末冶金材料科学与工程),2000,32(1):181-183
[28]CAO Du-Meng(曹笃盟).Thesis for the Doctorate of Central South University(中南大学博士论文).2006.
[29]LI Zhi-You(李志友),CAO Du-Meng(曹笃盟),ZHOU Ke-Chao(周科朝).Trans.Nonferrous Met.Soc.China(中国有色金属学报),2008,18(1):59-66
[30]Mishra K M,Lal A K,Haque F Z.Solid State Ionics,2004,167(1):137-146
[31]CAO Du-Meng(曹笃盟),LI Zhi-You(李志友),ZHOU Ke-Chao(周科朝).Journal of Functional Materials(功能材料),2004,35(z1):1791-1794
[32]WANG Gao-Jun(王高军),SU Guang-Yao(苏光耀),LI Zhao-Hui(李朝晖),et al.Chin.J.Power Sources(电源技术),2005,29(6):349-352
[33]Pistoia G,Pasquali M,Wang G,et al.Cheminform,1990,21(42):2365-2370
[34]QIAO Xiao-Ning(乔晓宁).Thesis for the Doctorate of Shaanxi University of Science and Technology(陕西科技大学博士论文).2016.
[35]SHI Chuan(石钏),REN Xiang-Zhong(任祥忠),ZHANG Pei-Xin(张培新),et al.The Proceedings of 29th the National Conference on Chemistry and Physics(第29届全国化学与物理电源学术年会论文集).Changsha:[s.n.],2011:98-99
[36]LI Hai-Xia(李海霞),JIAO Li-Fang(焦丽芳),YUAN Hua-Tang(袁华堂),et al.Chinese J.Inorg.Chem.(无机化学学报),2005,21(12):1865-1868
[37]ZHAO Bao-Xing(赵宝兴),HUANG Cheng-De(黄诚德),DONG Shu-Ben(董树本),et al.Chin.J.Power Sources(电源技术),2006,30(5):398-402
[38]Murphy D W,Christian P A,Disalvo F J,et al.J.Electrochem.Soc.,1981,128(10):2053-2060
[39]Gibbard H F.J.Power Sources,1989,26(1/2):81-91
[40]LIU Xiao-Jiang(刘效疆),LU Rui-Sheng(陆瑞生).Chin.J.Power Sources(电源技术),2002,26(1):26-28
[41]De Guibert A,Crepy G,Buchel J,et al.Proceedings of the 34th International.New York:IEEE,1990:145-147
[42]SHI Kui(石 奎),FENG Yu-Cai(冯 玉 才),ZHOU Ai(周 爱).Chin.J.Power Sources(电源技术),2016,40(9):1833-1835
[43]Hillel T,Ein-Eli Y.J.Power Sources,2013,229:112-116
[44]Guidotti R A,Reinhardt F W.Office of Scientificamp;Technical Information Technical Reports.Albuquerque:Sandia National Laboratories,1996.
[45]Dai J X,Lai M,Lafollette R M,et al.ECS Trans.,2011,33(27):3-9
[46]Dai J X,Lai M,Mckee J.220th Electrochemical Society Meeting:Vol.41.Honolulu:The Electrochemical Society,2012:11-16
[47]YAN Yan(闫妍),YANG Shao-Hua(杨少华),ZHAO Yan-Long(赵 彦 龙).Journal of Shenyang Ligong University(沈 阳理工大学学报),2014,33(2):6-8
[48]LUO Jian-Tun(骆柬氽),YANG Shao-Hua(杨少华),BAI Yin-Xiang(白银祥),et al.Chin.J.Power Sources(电源技术),2014,38(8):1516-1518
[49]DING Xiao-Ru(丁晓儒),YUAN Chao-Jun(袁朝军),YANG Shao-Hua(杨少华),et al.Journal of Shenyang Ligong University(沈阳理工大学学报),2016,1(35):102-105
[50]YUAN Chao-Jun(袁朝军),CAO Xiao-Hui(曹晓晖),YANG Shao-Hua(杨少华).Chin.J.Power Sources(电源技术),2015,39(11):2450-2452
[51]YUAN Chao-Jun(袁朝军),YANG Shao-Hua(杨少华),CAO Xiao-Hui(曹晓晖),et al.Chin.J.Power Sources(电源技术),2015,39(7):1479-1481
[52]YUAN Chao-Jun(袁朝军),YANG Shao-Hua(杨少华),CAO Xiao-Hui(曹晓晖).Journal of Functional Materials(功能材料),2015,46(17):17046-17048
[53]Wang F Y,Zhang H C,Liu L,et al.J.Alloys Compd.,2016,672:229-237
[54]SUN Rong(孙榕),YANG Shao-Hua(杨少华).Chin.J.Power Sources(电源技术),2017,41(2):270-271
[55]Cao J Q,Wang X Y,Tang A,et al.J.Alloys Compd.,2009,479(1):875-878
[56]SUN Rong(孙榕),YANG Shao-Hua(杨少华),CAO Xiao-Hui(曹晓晖),et al.Chin.J.Power Sources(电源技术),2016,40(11):2195-2197
[57]GUO Xue-Yi(郭学益),LIU Hai-Han(刘海涵),LI Dong(李栋),et al.Mining and Metallurgical Engineering(矿冶工程),2007,27(1):50-53
[58]Niu Y Q,Wu Z,Du J L,et al.Solid State Ionics,2014,255:80-83
[59]Guidotti R A,Reinhardt F W.IBA-2000 Manganese Oxide Battery Symposium:Vol.1.Albuquerque:Sandia National Laboratories,2001.
[60]Guidotti R A,Reinhardt F W.41st Power Sources Conference.Philadelphia:[s.n.],2004.
[61]Niu Y Q,Wu Z,Du J L,et al.J.Power Sources,2014,245:537-542
[62]Niu Y Q,Wu Z,Du J L,et al.Electrochim.Acta,2014,115:607-611
[63]Zhu S,Zhou H,Hibino M,et al.Adv.Funct.Mater.,2005,15(3):381-386
[64]MA Chun-An(马淳安),LOU Ying-Wei(楼颖伟),ZHAO Feng-Wu(赵峰呜),et al.Trans.Nonferrous Met.Soc.China(中国有色金属学报),2004,14(10):1736-1740
[65]Tarascon J M,Mckinnon W R,Coowar F,et al.J.Electrochem.Soc.,1994,141(6):1421-1431
[66]BAI Liang-Fei(白亮飞).Thesis for the Doctorate of University of Science and Technology of China(中国科学技术大学博士论文).2013.
[67]JIANG Jian-Bing(江剑兵).Thesis for the Doctorate of Central South University(中南大学博士论文).2014.
[68]YAO Yao-Chun(姚耀春).Thesis for the Doctorate of Kunming University of Science and Technology(昆明理工大学博士论文).2005.
[69]ZHENG De-Shan(郑德山),YANG Xiao-Ling(杨晓玲),YAO Xiao-Cui(姚小翠).New Chemical Materals(化工新型材料),2012,4:34-36
[70]Wang Z J,Du J L,Li Z L,et al.Ceram.Int.,2014,40(2):3527-3531
Development and Prospect of Oxide Cathode Materials for High Temperature Lithium Batteries
SONG Heng-Xu1NIU Yong-Qiang1HOU Hua1WU Zhu2ZHAO Yu-Hong*,1
(1Laboratory of Materials for High Temperature Lithium Batteries,School of Materials Science and Engineering,North University of China,Taiyuan 030051,China)
(2Energy Science and Technology Laboratory,Shanghai Institute of Microsystem and Information Technology,Chinese Academy of Sciences,Shanghai 200050,China)
High temperature lithium battery is the development of low temperature applications of thermal batteries,and has large potential development and utilization in the oil/gas and geothermal field.Compared with the lithium alloy anode materials with large specific capacity and close to the pure lithium electrode potential,the cathode materials are pivotal section of high temperature lithium battery and have large potential space.In the cathode materials,the oxide materials exhibit high voltage characteristics and high thermal stability,and the high-temperature lithium battery can be miniaturized to meet the current voltage supply under the specific conditions.However,there is no systematic overview about the oxide of high-temperature lithium battery currently.In order to make more development in this field and promote the optimal adjustment of energy structure,this paper introduces and summarizes the transition metal oxides used in high-temperature lithium battery,including physical properties,electrochemical properties and synthesis or preparation methods.We describe the excellent properties of various oxide materials in detail,and point out the shortcomings and the existing problems of the material.Moreover,this paper predicts the development of oxide cathode materials,anddemonstrates the future direction of development and the work to be done.We hope that this work can provide some reference for the relevant researchers.
high-temperature lithium battery;cathode material;oxide;electrochemical performance;preparation
TM911.16
A
1001-4861(2017)12-2155-14
10.11862/CJIC.2017.263
2017-07-24。收修改稿日期:2017-09-10。
国家自然科学基金(No.21473234,51774254)和中北大学校青年基金(No.110246)资助项目。
*通信联系人。 E-mail:zhaoyuhong@nuc.edu.cn
猜你喜欢
蓄电池(2022年1期)2022-02-25
陶瓷学报(2020年6期)2021-01-26
中学生数理化·中考版(2018年11期)2019-01-31
电子制作(2018年23期)2018-12-26
教学考试(高考化学)(2018年5期)2018-12-06
科技知识动漫(2017年4期)2017-04-15
中国有色冶金(2016年3期)2016-02-11
百科探秘·航空航天(2015年3期)2015-12-01
科技资讯(2015年8期)2015-07-02
储能科学与技术(2014年5期)2014-02-27
