
固相萃取-超高效液相-串联质谱法检测水产品中渔药残留
2017-11-15 10:52:59张海霞王迎迎
黑龙江水产 2017年5期
张海霞 王迎迎
(哈尔滨市农产品质量安全检验检测中心 黑龙江 哈尔滨 150070)
固相萃取-超高效液相-串联质谱法检测水产品中渔药残留
张海霞 王迎迎
(哈尔滨市农产品质量安全检验检测中心 黑龙江 哈尔滨 150070)
在水产品养殖过程中,由于使用者对氯霉素、磺胺类、喹诺酮类渔药及三苯甲烷类染料的毒性和危害认识不足,存在违规使用的现象.渔药残留常见危害为药物通过较长时间的蓄积,对人体产生的慢性毒性作用.人们长期食用这类含渔药残留的水产品后,药物在人体内蓄积,达到一定浓度后,就会对人体产生各种慢性毒性作用,损害肝脏、肾脏、消化系统、神经系统、造血系统、循环系统等.农业部235号公告规定了其限量标准,农业部和黑龙江省农监局每年要对水产品中这四类渔药的残留进行四次例行监测,以保证水产品的质量安全.目前,水产品中渔药残留的检测方法主要有酶联免疫吸附法、高效液相色谱法、气相色谱法、气相色谱-串联质谱法、液相色谱-串联质谱法、电化学分析、毛细管电泳等,使用的前处理方法主要有液液萃取法、固相萃取法等.这些方法对渔药残留的检测多是按同族药物或单一药物来开发,这些方法的不足之处在于仅可以检测单一或几个目标化合物,检测效率低,成本高,造成检测仪器设备、检测人员的浪费.本实验采用Prime HLB固相萃取柱净化,超高效液相色谱-串联质谱法一次性检测4类12种渔药残留,检测效率高,成本低,适用于越来越繁重的突发事件和应急监测工作.
1 材料与方法
1.1 仪器、试剂与材料
仪器:超高效液相色谱-串联四级杆质谱联用仪Waters Acquity UPLC-TQD(美国Waters公司),配有电喷雾离子源;Milli-Q高纯水发生器(美国Millipore公司);高速离心机(Sigma);高速组织匀质仪、涡旋振荡器(IKA);氮吹仪(美国Organomation);pH计、电子天平(Mettler Toledo);超声波清洗器(KUDOS);料理机(博朗);试剂:优级纯的乙腈、甲酸、氯化钠、盐酸羟胺、对甲苯磺酸、无水乙酸钠、冰乙酸(科密欧);乙腈、甲醇色谱纯(美国Fisher公司);甲酸色谱级(迪马);Prime HLB固相萃取柱(Waters);标准品均购自Dr.Ehrenstorfer.样品:供试样品鲤鱼,购自当地超市.鱼去鳞,取脊背上的鱼肉,切成小块,在料理机中绞碎.
1.2 样品的提取
称取5.0g绞碎的鱼肉样品于50mL具塞离心管内,加入混合内标工作溶液,涡旋混匀.加入1.5mL20%的盐酸羟胺溶液,2.5mL1.0mol/L的对甲苯磺酸溶液,5.0mL乙酸盐缓冲溶液,乙腈8mL,涡旋混匀后,超声5min,再加入8g氯化钠,涡旋混匀60s,7000r/min离心5min,收集上清液的乙腈层于50mL烧杯中.再向离心管中加入乙腈8mL,涡旋混匀60s,7000r/min离心5min,合并乙腈层的提取液.
1.3 样品的净化
Prime HLB固相萃取柱,加入与提取液pH值相近的甲酸乙腈1mL进行固相萃取小柱的活化.将50mL烧杯中的提取液转移至固相萃取小柱,收集全部流出液,40℃下氮吹至近干.用200µL乙腈溶解后,800µL水定容至1mL,过0.22µm的滤膜.供超高效液相色谱-串联质谱测定.
1.4 仪器条件
超高效液相色谱条件:色谱柱: Waters ACQUITY UPLC BEH C18柱(1.7µm,2.1x50 mm );柱温: 35℃; 进样体积: 5µL;流速: 0.3mL/min;流动相1:A为乙腈, 流动相B为0.1% 甲酸溶液.具体洗脱程序:0~2.0min10~25%A,2.0~3.0min25~45%A,3.0~4.0min45~90%A, 4.0~5.5min90%A,5.6~7.0min10A.
质谱条件:离子源: 电喷雾电离源( ESI),正、负离子模式;源温度: 110℃;毛细管电压:+0.5kV,-2.0 kV;脱溶剂温度:400℃;脱溶剂气流速(N2):800L/h;锥孔气速: 50L/h.采用MRM多反应检测模式进行检测.
2 结果与分析
2.1 提取方法
水产品中磺胺类、喹诺酮类和氯霉素的提取试剂主要有乙酸乙酯、甲醇、不同酸度的乙腈等,而孔雀石绿的提取采用了乙腈-盐溶液或乙腈作为提取试剂[12].本实验对比了乙腈和乙腈-盐溶液对鱼肉中的12种化合物的提取效率,结果表明,在使用单一的乙腈提取时,孔雀石绿、隐色孔雀石绿及其内标物的提取效率为零,没有任何回收,其余的化合物的回收较好.采用乙腈-盐溶液为提取试剂,在保证了磺胺类、喹诺酮类和氯霉素的提取效率的前提下,孔雀石绿和隐色孔雀石绿的回收率能满足要求.水产品中渔药残留的提取主要有高速组织匀质、超声波提取、震荡提取,本实验对以上三种提取方法进行对比.高速组织匀质机进行匀质提取时,由于鱼肉较粘,会大部分样品粘黏在匀质机的刀头上,降低提取效率.震荡和超声波提取都能达到较好的提取效果,但震荡提取时离心管是横放在振荡器上的,提取液容易从离心管盖溢出,综合考虑提取效率及可操作性,选取超声波提取.
2.2 样品净化
基质效应是影响LC-MSMS定量分析准确性的重要因素.样品净化效果不好会影响化合物的离子化,基质效应强,导致色谱峰峰型不好,并且对色谱柱和质谱有污染.基质效应通过对每个待测物的基质增强或抑制进行评价.具体的计算公式为ME=Rm/Rsⅹ100%,其中Rm为目标化合物在空白基质溶液中的响应值,Rs为目标化合物初始流动相溶液中的响应值[13].固相萃取柱的基本操作步骤为活化、上样、淋洗、洗脱.Prime HLB固相萃取柱的工作原理是吸附提取液中的杂质已达到净化的目的,产品的使用方法是不用活化即可使用.为了验证不活化的固相萃取柱的去除杂质的效果,比对了固相萃取柱不活化、样品空白提取液活化和pH=1.40的甲酸乙腈(与提取液中的pH值相近)活化三种不同的方式.以基质效应的强弱来衡量不同活化方式的净化效果.三种不同活化方式的基质效应比较见表1.由表可见,除隐色孔雀石绿为基质减弱效应外,其余参数的三种活化方式均为基质增强效应,其中样品空白提取液活化和pH=1.40的甲酸乙腈活化的基质效应差异不明显.使用样品提取液进行活化,需要每次检测前都找到阴性样品,增加工作量和工作难度,最终本实验选取pH=1.40的甲酸乙腈为活化试剂.
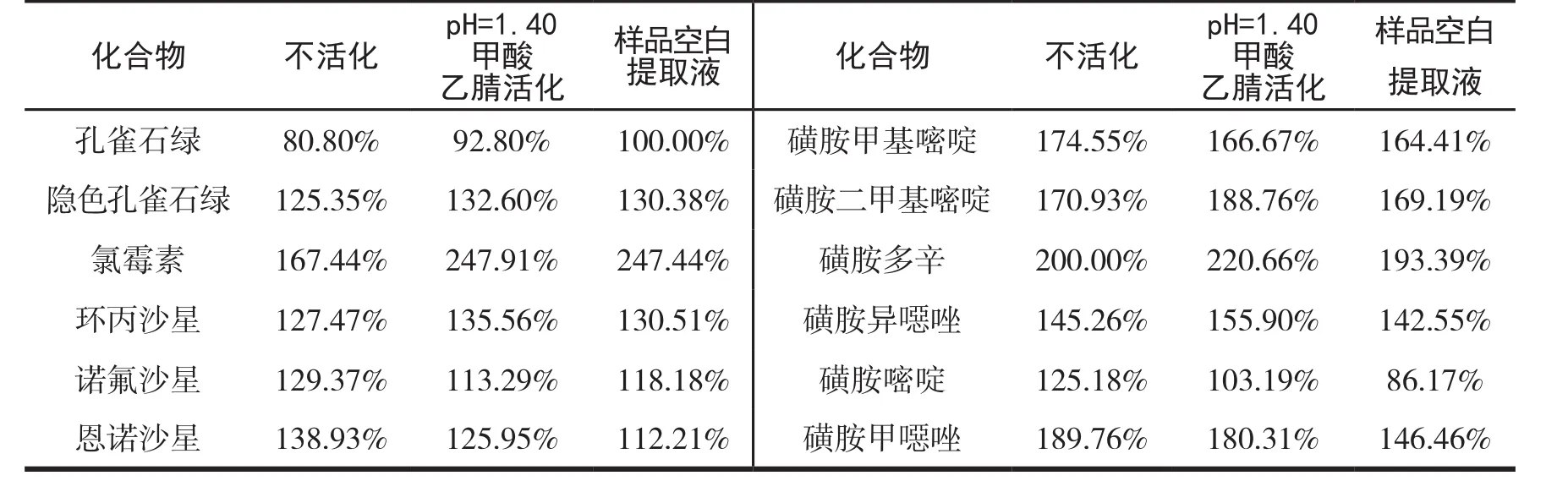
表1 三种不同活化方式的基质效应对比
2.3 质谱方法的优化
配制浓度为1mg/kg的12种渔药及其内标物的标准溶液,以流动注射方式连续进样,进行母离子扫描.按照欧盟(2002/657/EC)指令规定,低分辨率质谱联用检测应确定在母离子的基础上选择两个以上子离子.在确定各个化合物母离子后,对其母离子进行碰撞扫描,每个化合物再选择2个响应值高的特征离子对作为定量及定性离子,同时对其进行MRM参数优化.12种渔药及其内标化合物中只有氯霉素及其内标物为ESI-模式,其余的参数均为ESI+模式.优化的质谱参数见表2.各化合物的总离子流图见图1.
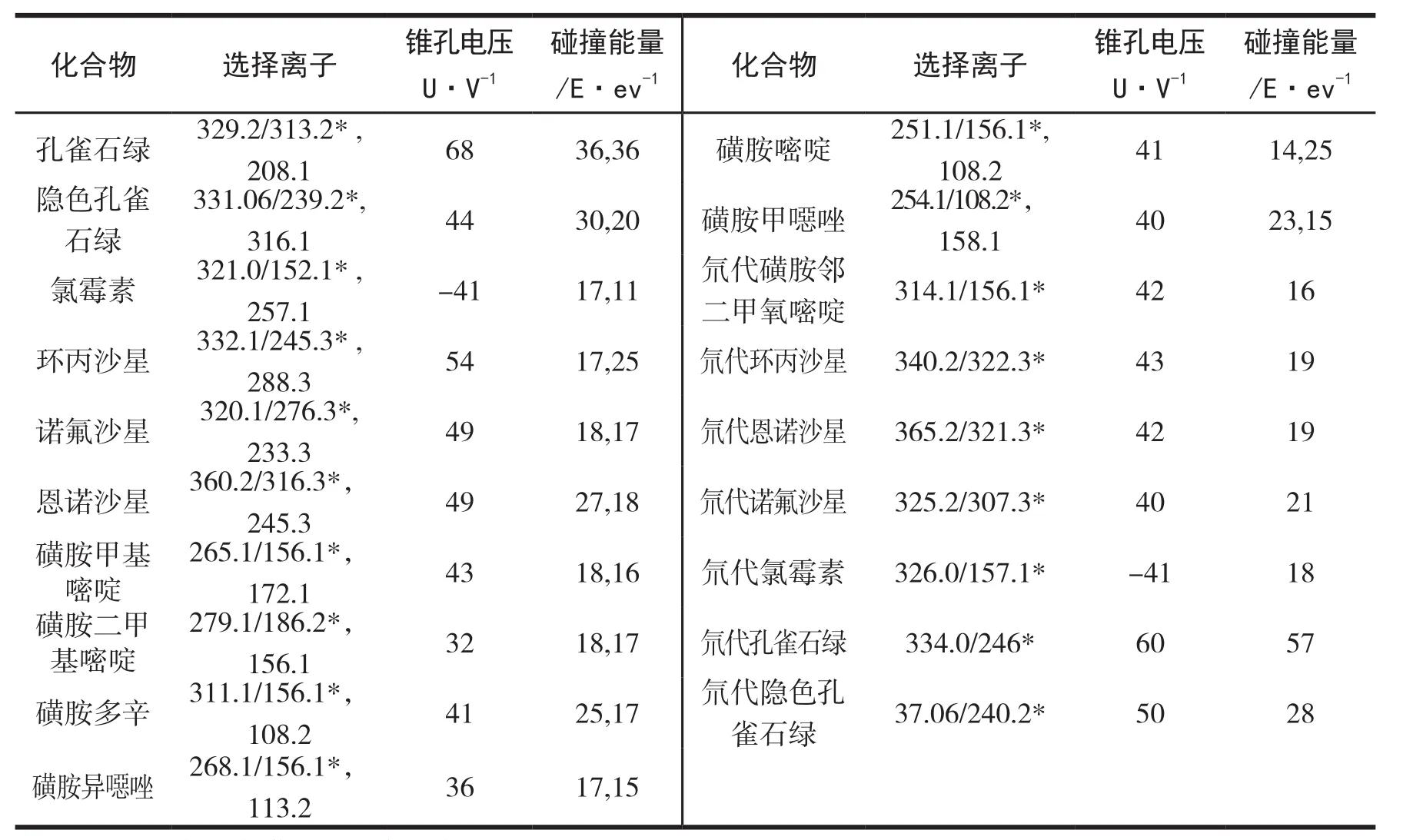
表2 12种渔药及内标标品多反应监测扫描模式(MRM)的质谱参数

图1 12种化合物及其内标物的总离子流图
2.4 液相方法的优化
由于这12种化合物及其内标物的化学性质差异较大,在质谱中采用了正离子和负离子的采集模式,为了使每个化合物能够更好的离子化,需要对使用的流动相体系进行优化.根据查阅的文献及本实验室的工作经验,对这12种化合物仪器分析时所采用有机相主要有乙腈、甲醇;水相主要有水、甲酸-水、乙酸铵.本实验进样适当浓度的混合标准品,对乙腈-5mmol/L乙酸铵、乙腈-0.1%甲酸、甲醇-水三种流动相进行优化.各待测物在不同流动相体系中的响应值区别见图2.由图2可以看出,以乙腈-5mmol/L乙酸铵为流动相时氯霉素的响应值高,但喹诺酮化合物的响应值低,其中环丙沙星没有响应;以甲醇-水为流动相时磺胺类化合物没有响应或响应值低;以乙腈-0.1%甲酸为流动相时,氯霉素的响应值较以乙腈-5mmol/L乙酸铵为流动相低,但能满足检测需求,其他化合物的响应值均较高,也在检测需求范围内.最终选择以乙腈-0.1%甲酸为流动相.

图2 待测物在不同流动相体系中的响应值
2.5 方法的线性、检出限和定量限
为了消除基质效应对定量结果造成的影响,以不含待测物的空白样品为基质液,配制成基质标准溶液,磺胺类、氯霉素、喹诺酮类化合物浓度分别为10、50、100 、200、300、500、1000µg/mL,孔雀石绿和隐色孔雀石绿的浓度分别为5、10、15 、20、25、50、100µg/mL,进行方法线性的测定.以待检参数的面积(y)为纵坐标,质量浓度(x)(mg/L)为横坐标,建立标准曲线,得到线性回归方程.线性范围及相关系数见表3.以目标化合物在空白基质中的信噪比(S/N)来获得检出限和定量限,S/N=3时对应的含量为检出限(LOD),S/N=10时对应的含量为定量限(LOQ).

表3 12种渔药的线性范围、线性方程及相关系数
2.6 方法的回收率、精密度
按照不同化合物的检出限和国家规定的限量标准,每种化合物在样品中添加低、中、高三种水平浓度的标品,每个水平重复6次,按照2.2的试验方法进行处理,计算方法的回收率和精密度.具体的添加浓度、回收率和精密度见表4.回收率在70.3-118.7%之间,精密度为1.5-6.7%,符合国家检测标准的要求.

表4 方法的添加回收率和精密度(n=6)
3 结论
本实验采用乙腈-盐溶液提取,Prime HLB固相萃取柱净化,超高效液相色谱-串联质谱分析,建立了能一次性检测水产品中磺胺类、喹诺酮类、孔雀石绿以及氯霉素四类12种渔药残留的方法.通过对方法的检出限、回收率、精密度、线性范围等方法学进行验证,该方法满足对这12种渔药检测的要求.与传统的方法相比,本实验的方法快速、简单、灵敏、成本低、效率高,适合实验室进行批量样品的检测.
略)
张海霞 性别:女 职称:高级农艺师 电话:15204666138 Email:zhanghaixianj@163..com
猜你喜欢
当代水产(2021年11期)2022-01-15 05:39:14
当代水产(2021年9期)2021-12-02 01:34:42
科教导刊·电子版(2021年1期)2021-03-28 03:31:54
当代水产(2019年8期)2019-10-12 08:57:48
当代水产(2019年5期)2019-07-25 07:50:22
环境与发展(2019年11期)2019-02-12 12:35:02
山东化工(2019年1期)2019-01-24 03:00:16
中国蜂业(2018年4期)2018-05-09 06:25:08
当代化工研究(2016年6期)2016-03-20 16:21:46
无机化学学报(2014年3期)2014-02-28 17:30:58
