
辣椒(Capsicum annuum L.)果实发育及品质相关新microRNA的鉴定与分析
2017-11-10 03:23:49LIUZhoubin,ZHANGYuping,OULijun等
辣椒杂志 2017年3期
辣椒(Capsicum annuum L.)果实发育及品质相关新microRNA的鉴定与分析
LIU Zhoubin1,2,aZHANG Yuping3,aOU Lijun2,aKANG Linyu1,2LIU Yuhua1,2LV Junheng1,2WEI Ge2YANG Bozhi2YANG Sha2CHEN Wenchao2DAI Xiongze2LI Xuefeng2ZHOU Shudong2ZHANG Zhuqing2,*MA Yanqing2,*ZOU Xuexiao1,2,**
(1. 湖南大学研究生院隆平分院, 长沙 410125; 2. 湖南省蔬菜研究所, 长沙 410125;3. 湖南农业大学资源环境学院, 长沙 410128; a为共同第一作者)
MicroRNA(miRNA)是一种非编码的小RNA,在各种生物过程中起着重要的调节作用。前人研究表明,模式植物中miRNA与果实发育相关。然而,人们对辣椒(Capsicum annuum L.)果实品质及发育相关的miRNA了解尚不清楚。本研究利用二代测序技术(Next-generation sequencing technology)比较了不同品种辣椒果实在不同发育时期的小RNA。利用miRDeep2软件从4个样本中共鉴定出了59个已知miRNA和310个新miRNA。预测了新miRNA的靶基因,共656个,并对其中402个靶基因进行了注释。GO分析和KEGG途径表明,某些靶基因与淀粉糖、氨基糖以及核糖代谢相关。qPCR表明一些miRNA的表达模式与其靶向基因相反,该结果为今后研究miRNA调控果实发育和品质形成提供了重要基础。
辣椒(Capsicum annuum L.);miRNA;果实发育;果实品质;深度测序;
1 引言
辣椒(Capsicum annuum L.)属于茄科植物,是种植范围最广、最重要的蔬菜作物之一。其果实具有特殊的颜色、辛味和香气,因此常被作为调味品使用[1]。辣椒果实能够积累辣椒素、色素(花青素和胡萝卜素)和维生素A、B、C[2]。一个世纪以来,在辣椒的育种中,产量和果实品质是重要的质量参数[3]。果肉厚度、总可溶性固形物是许多植物果实品质的重要特征[4]。Isabelle 等[5]研究表明,辣椒果实能积累大量的维生素C。相比成熟的果实,处于成熟期的果实维生素C含量更多,还原糖更少[6]。近年来,为研究辣椒果实发育和辣椒素合成,辣椒属转录组得到了分析。Martinez-Lopez等[7]于2014年研究分析了墨西哥辣椒(“Tampiqueño 74”)花后(DAA)10、20、40、60天的果实转录组数据。RNA-seq结果表明,辣椒素和抗坏血酸生物合成的相关基因在果实成熟过程中大量表达[7-8]。
microRNA是一类长度约为21个核苷酸(nt)大小的非编码小RNA,在植物发育、信号转导、生物和非生物胁迫响应等生物过程中起着重要作用[9]。通常情况下,单链小RNA在经DICER-LIKE1酶(DCL1)的处理后,形成miRNA复合物[10]。复合物展开后,成熟的单链miRNA得到释放。最后,成熟miRNA通过互补转录的分裂或翻译阻遏作用进入到RNA诱导的沉默复合体(RNA- induced silencing complex,RISC)中[11]。目前在一些模式植物中已经发现了许多miRNA,这些miRNA及其靶基因的发现使人们有可能进一步了解miRNA介导的基因调控网络和生物机制。近来辣椒基因组已经得到测序[12-13],这为今后辣椒miRNA的研究提供了基础。已经有研究利用高通量测序技术,从辣椒中鉴定出29个保守的miRNA家族和35个新的miRNA家族[14]。然而,miRNA在辣椒果实发育和品质形成中的作用尚不明确。
本研究使用Illumina HiSeq 2500平台研究了辣椒果实在成熟过程中品质相关的miRNA的表达情况。鉴定了与果实成熟相关的差异表达miRNA,并预测了相关的靶基因。随后对差异表达的miRNA及其靶基因的功能进行了讨论。此外,还利用qPCRPCR检测了一些miRNA及其靶基因的表达模式。这些数据为研究辣椒果实发育及品质形成的分子机制提供了新的视角。鉴定和分析已知及未知的miRNA有助于更好地理解miRNA在辣椒果实发育中的作用。
2 材料和方法
2.1 植物材料
于湖南省蔬菜研究所,以正常条件(16h/8h,26℃/20℃)栽培两个辣椒品种“Luosijioa”和“06J19-1-1-1-2”。 “Luosijiao”在中国境内广泛种植,其口味和维生素C含量优于“06J19-1-1-1-2”。在开花时对“Luosijiao”植株进行标记,取绿熟期果实(30DAA, A1)、转色期果实(40DAA, A2)和红熟期果实(50DAA, A3)。对于“06J19-1-1-1-2”品种,取花后30天绿熟期的果实(30DAA, B1)。每个时期的样品从多株上取样并混合,-80℃保存。
2.2 RNA提取与深度测序
根据说明书,使用Trizol(Invitrogen)提取不同时期辣椒果实的总RNA。每个样品总RNA取5 μg建小RNA库。使用NEB Next Multiplex Small RNA Library Prep Set for Illumina ( NEB, 美国 )试剂盒,按照说明书构建测序用库。使用T4 RNA连接酶为小RNA加上3’末端和5’末端的接头,随后使用Super-Script II Reverse Transcriptase (RNase H-)试剂盒(Invitrogen)将RNA反转录成单链cDNA。之后使用LongAmp Taq2X Master Mi和相应引物对上述反应物进行PCR扩增。PCR产物使用8%聚丙烯酰胺凝胶纯化(100 V, 80 min)。使用高灵敏的DNA芯片和Aglilent 2100生物分析仪进行质量评估后,回收140~160 bp长度的DNA片段,并溶解于8 μL的洗脱缓冲液中,用于测序。4个样品由Biomarker Technologyies公司(北京)使用Illumina HiSeq 2500测序平台进行单末端(SE)50 nt测序。
2.3 miRNA的生物信息学分析
对测序原始数据(raw data)进行清洗,去除低质量的序列,如:接头、低于18 nt的序列、多N序列等。在RepeatMasker和Rfam数据库(http://www.sanger.ac.uk/software/Rfam)中对清洁数据(clean data)进行blast操作,去除rRNA、tRNA、snRNA、snoRAN及其他非编码RNA。剩余的序列置于miRBase 21.0数据库(http://www.mirbase.org)中搜索,以鉴定已知的miRNA。按Friedlaender等[15]的研究方法,将没有注释的sRNA序列使用bymiRDeep2软件通过检测发卡结构、Dicer酶切位点和最低自由能的方法预测新miRNA。新miRNA的标准采用Meyers等人[16]的研究结果。
2.4 miRNA的差异表达分析
为定义miRNA的表达水平,将miRNA的表达量进行TPM校正,公式如下:标准表达量=比对的序列数/总序列数×106。使用R语言的DEGseq包分析2个样品间不同的表达水平,使用q值对p值进行校正[17]。q < 0.01 和 │log2 (fold change)│> 1为差异表达显著的指标。
2.5 靶基因预测与富集分析
本研究基于辣椒转录本的序列相似性,使用psRNATarget分析软件预测miRNA[12,18]。E值设为10-5,在KOG(Clusters of Orthologous Groups of proteins)、KEGG(Kyoto Encyclopedia of Genes and Genomes)和GO(Gene Ontology)三个数据库中进行BLAST搜索,对功能和途径进行分类。使用R语言的TopGO包进行GO富集分析,FDR <0.05则认为显著富集[19]。富集的基因使用WEGO进行绘图(http: //wego.genomics.org.cn/ cgi-bin/ wego/index/ pl)。
2.6 定量RT-PCR(qRT-PCR)和stem-loop qRT-PCR
使用Trizol(Takara,大连)提取样品总RNA,并使用RNase-free Dnase I (Promega,美国)对总RNA进行处理。对于miRNA,取2 μg经DNase I处理的总RNA,使用Prime-Script RT reagent Kit(Takara,中国大连)进行反转录。反转录反应条件如下:16 ℃孵育 30 min;60 个循环:30℃ 30 s,42℃30 s,50℃ 1 s;70℃ 5 min[20]。对于靶基因,取 1 μg经DNase I处理的总RNA,使用Prime-Script RT reagent Kit(Takara,大连)和oligo (dT) 18引物进行合成。反转录反应条件如下:37℃ 15 min,42℃ 1 h,4℃保存。所有引物于表S1中列出。
以FastStart Universal SYBR Green Master Mix(Roche)为染料,在StepOne plus PCR(Applied Biosystems)平台上进行miRNA和靶基因的实时荧光定量PCR分析。qRT-PCR反应条件如下:95℃ 10 min,40个循环:95℃ 15 s,56℃ 30 s,72℃ 15 s。CpAction作为内参基因。使用溶解曲线检测产物的特异性。所有qRT-PCR反应均设置3个生物学重复,并使用2-ΔΔCt法对相对表达水平进行校正[21]。
2.7 支撑数据获取
本研究中使用的所有测序数据均可从SRAArchive(http://www.ncbi.nlm.nih.gov/sra)获得,登记号为:SRP076911。
3 结果
3.1 小RNA库测序概述
为鉴定辣椒果实发育和品质形成相关的miRNA,利用深度测序构建了3个“Luosijiao”小RNA库,1个“06J19-1-1-1-2”库。4个库中共计83 639 907个序列。去除接头、低质量序列、小于18 nt序列和多A序列后,清洁数据数量在16 365 419(A3)至 22 799 181(A1)之间(表 1)。4个样品分别有 16 075 461、11 662 314、9 236 660和12 481 144个序列特异性地比对到了基因组上。本研究几乎检测了每个类型的RNA,包括rRNA、snRNA、snoRNA、tRNA和重复序列相关的sRNA(表1)。并且使用miRDeep 2软件对已知miRNA和新miRNA进行了预测(表1),未注释的序列用于之后的分析。
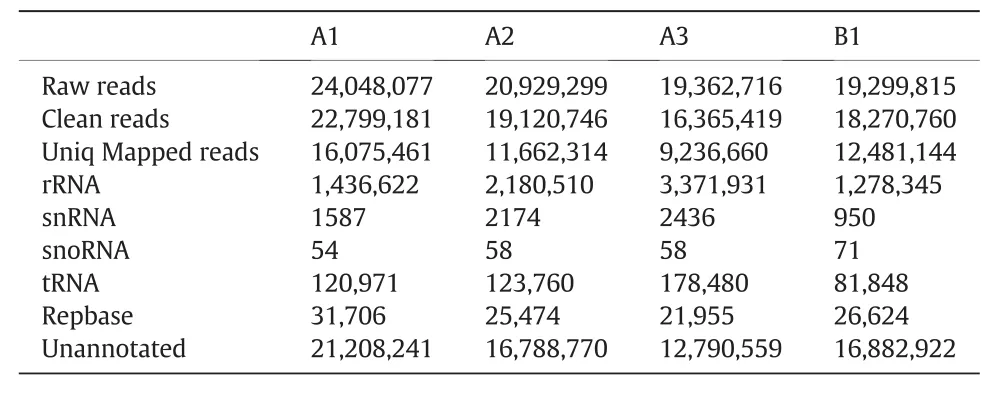
表1 不同种类小RNA的分布情况
总体来看,除去各库间相同、不同的sRNA,临近库中约30%的sRNA相同,特异性序列仅有少部分(9.73%~11.16%)(图1)。
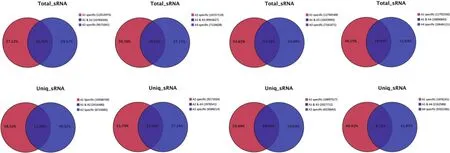
图1 不同样品间共有或特有的小RNA序列
3.2 已知及新miRNA的特征
4个样本中,小RNA(sRNA)序列长度在18~ 28 nt之间,大部分在20 ~ 24 nt之间。其中24 nt的sRNA数量最大,约占4个样品的20% ~ 35%;紧随其后的是23 nt的sRNA,从长度来看是典型的miRNA(图1A)。
Kudla等[22]研究表明碱基组成会影响miRNA的理化性质、生物学特性及其二级结构。拟南芥中,不同的Argonaute蛋白(AGO)在不同的miRNA起始位置富集。AGO1偏爱以U起始的miRNA,而AGO2和AGO4偏爱5’末端以A为结尾的miRNA[23]。miRNA对核酸的偏好表明,约有50%的miRNA的第1个核苷酸是A(图2B)。从长度来看,以U为开头的miRNA长度分别为18 nt、21 nt、22 nt,其余miRNA以A开头(图2C)。
3.3 辣椒已知miRNA和新miRNA的鉴定
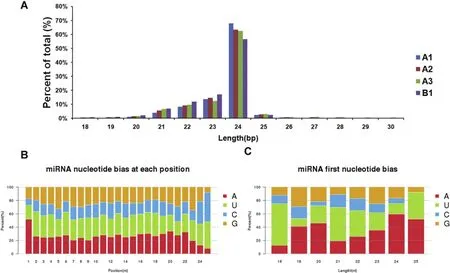
图2 辣椒4个样品中的小RNA
经过数据过滤,将数据与miRBase 20.0库(http://www.mirbase.org)中的成熟miRNA及前体miRNA对比后,鉴定出59个已知miRNA家族(表2)。此外,鉴定了12个已知miRNA的靶基因,共36个且大多数在植物中保守(表S3)。除已知miRNA外,将剩余的序列被比对到辣椒基因组,用于鉴定新miRNA。在miRBase 20.0中没有比对到保守序列且没有报道过的miRNA即视为新miRNA。通过miRDeep2软件分析,共从4个样品中得到310个新miRNA(表S4)。为验证这些sRNA序列是否为辣椒果实的miRNA,使用byMfold或RNAfold软件检测其发卡结构[24]。结果表明,这些miRNA的前体均含有特异的茎环结构(表S4和图S1)。4个新miRNA的二级结构如图3所示。所有新miRNA的表达水均较低。此外,辣椒新miRNA的异构体也被鉴定出来,共有10个异构体(表S5)。
3.4 靶基因的GO注释和KEGG途径分析
Jones-Rhoades[25]、Hu等人[26]的研究结果表明,miRNA与靶标mRNA的结合几乎完美地基于序列互补,某种程度上类似RNA干扰。该现象为预测miRNA的靶基因提供了有效的方法,即比较miRNA和mRNA的序列即可。本研究共鉴定出310个新miRNA,预测了656个靶基因,其中402个得到了注释(表S6)。此外,对靶基因进行了GO分析,用于功能分类。GO富集分析表明“代谢过程”、“生物调节”、“结合”、“催化活性”是相生相伴的关系(图4、表S7)。KEGG途径分析表明,许多miRNA与不同的代谢途径相关。有趣的是,“Luosijiao”和“06J19-1- 1-1-2”的氨基糖代谢、核糖代谢以及淀粉和蔗糖代谢丰富,表明某些miRNA参与了果实品质形成(表S8)。
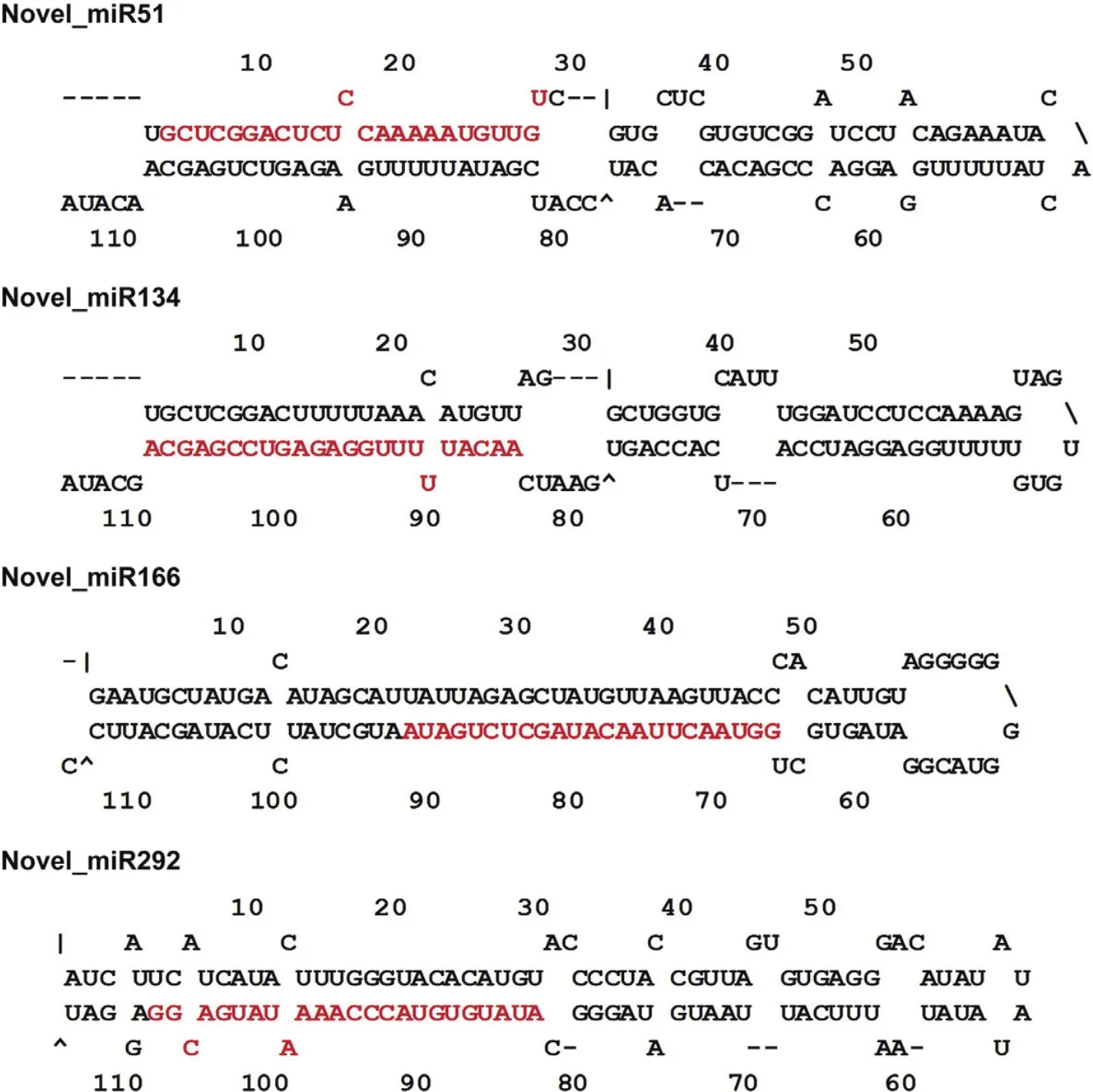
图3 新miRNA二级发卡结构的预测
3.5 差异表达分析
为深入理解不同辣椒果实样品中miRNA的表达模式,我们计算了每个样品的RPKM值。结果表明miRNA在果实发育过程中表达水平下调(A1、A2、A3),不同品种同一发育阶段的表达水平具有可比性(A1、B1)(图5A)。同时我们使用本实验室未发表的转录组数据(SRP075936)比较了miRNA及其靶基因的表达谱,发现大多数已知miRNA及其靶基因的表达模式呈负相关的关系(图5B和表S3)。为验证miRNA及其靶基因的表达模式,分别挑选了4个已知和4个新miRNA及其对应的靶基因进行qRT-PCR分析(图6)。结果表明,多数miRNA与其靶基因的表达水平呈负相关。3个 miRNA(can-miR156a、canmiR160a和 canmiR396a-5p)在果实DAA50时期(A3)高度表达,而其靶基因下调表达(图6A)。多数新miRNA的表达水平偏低(图6B)。然而,其表达仍会影响靶基因的表达水平(图6B),表明这些新miRNA在辣椒果实发育中仍然发挥了作用。
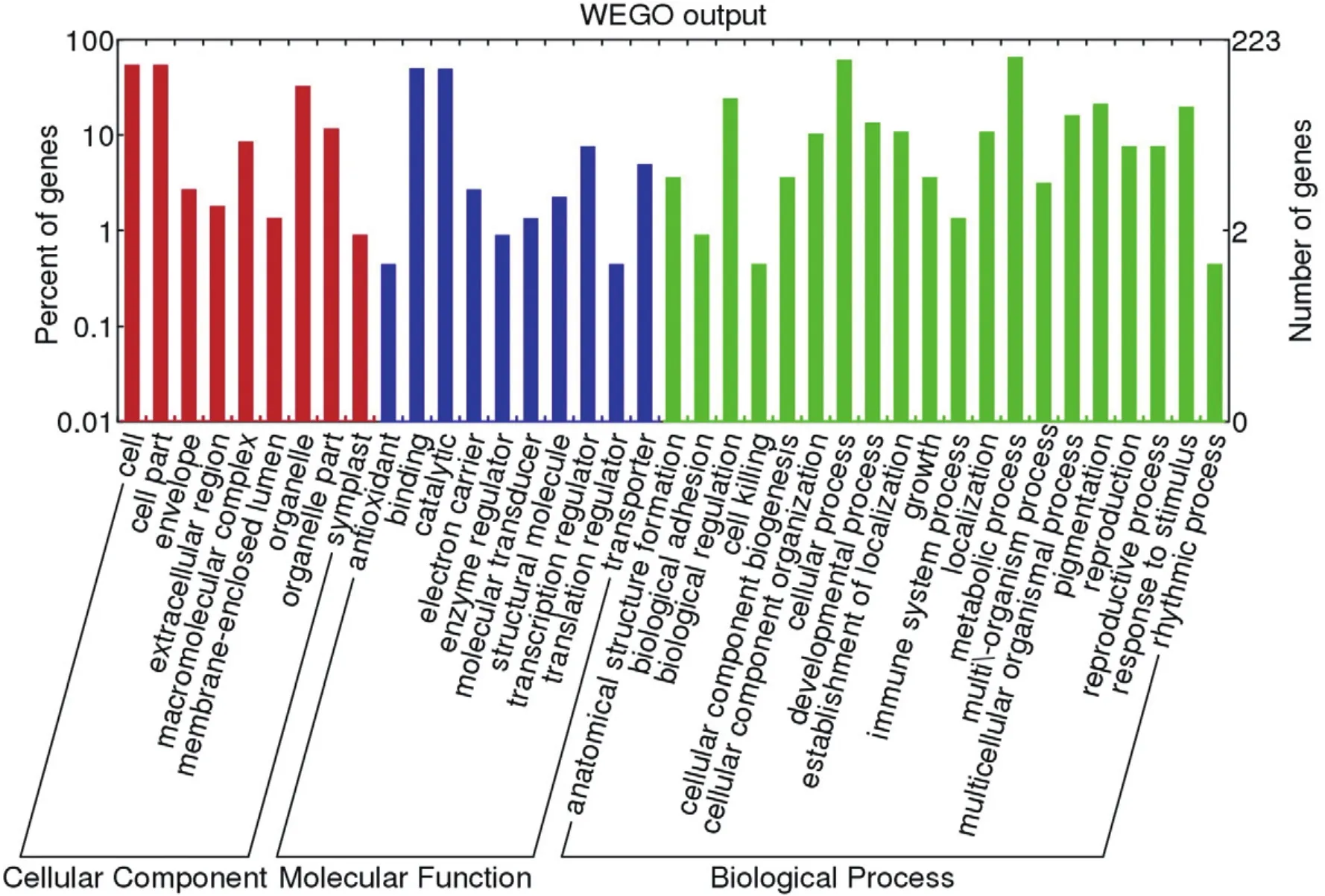
图4 靶基因的GO分类
4 讨论
尽管我们在植物果实发育研究中取得了一定进展,但是对辣椒果实发育及品质形成的调控网络理解尚浅。由于非编码RNA、miRNA在各种发育和生物过程的转录后水平上具有不尽相同的重要作用[9,11],我们使用小RNA高通量进行测序,比较了不同辣椒品种在不同果实发育阶段的miRNA表达谱,鉴定出了59个已知miRNA和310个新miRNA。
miRNA参与了植物体内多种细胞、生理及发育过程的调控。在植物中,某些miRNA具有高度保守的序列和功能。miRNA多通过靶向裂解mRNA来发挥调控作用,因此miRNA对mRNA的积累起负调控作用,两者的表达模式在同一植物细胞中呈负相关的关系。miR156调控一系列Squamosapromoter binding protein-like(SPL) 基 因[27-28]。Zhang[23]、Ferreira E Silva 等人[29]在番茄中对 slymiR156a和At-miR156b进行超表达后,果实分别表现出多心皮和结构异位的性状。此外,Sun等[30]在拟南芥中进行Md-miR156h基因的异源表达后,角果出现长度缩短和部分不育的情况。本研究中,多数属于can-miRNA家族的miRNA在4个样品中的表达具有差异(图5B、图6A)。can-miR156基因在DAA50(A3)中上调表达,在06J19-1-1-1-2的果实中(A1)下调表达,表明can-miR156与辣椒果实发育和品质形成有关。
生长素响应因子(auxin response factor,ARF)是一类调节生长素信号转导的转录因子。Hwang[14]等2013年研究表明ARF是can-miR160和can-miR160家族的靶基因。本研究中,3个ARF 基因中,ARF10(Capana11g000076)、ARF17(Capana04g001778)和 ARF18(Capana06g000514)同样被预测为can-miR160的靶基因(表S3)。并且验证了can-miR160a和靶基因ARF18的表达谱(图5B、图6A)。miR167和miR160调节植物细胞对生长素的响应,在果实发育中具有重要作用[31-32]。F-box蛋白家族是miR394的靶基因,在一些植物激素信号转导途径中具有重要作用[33]。
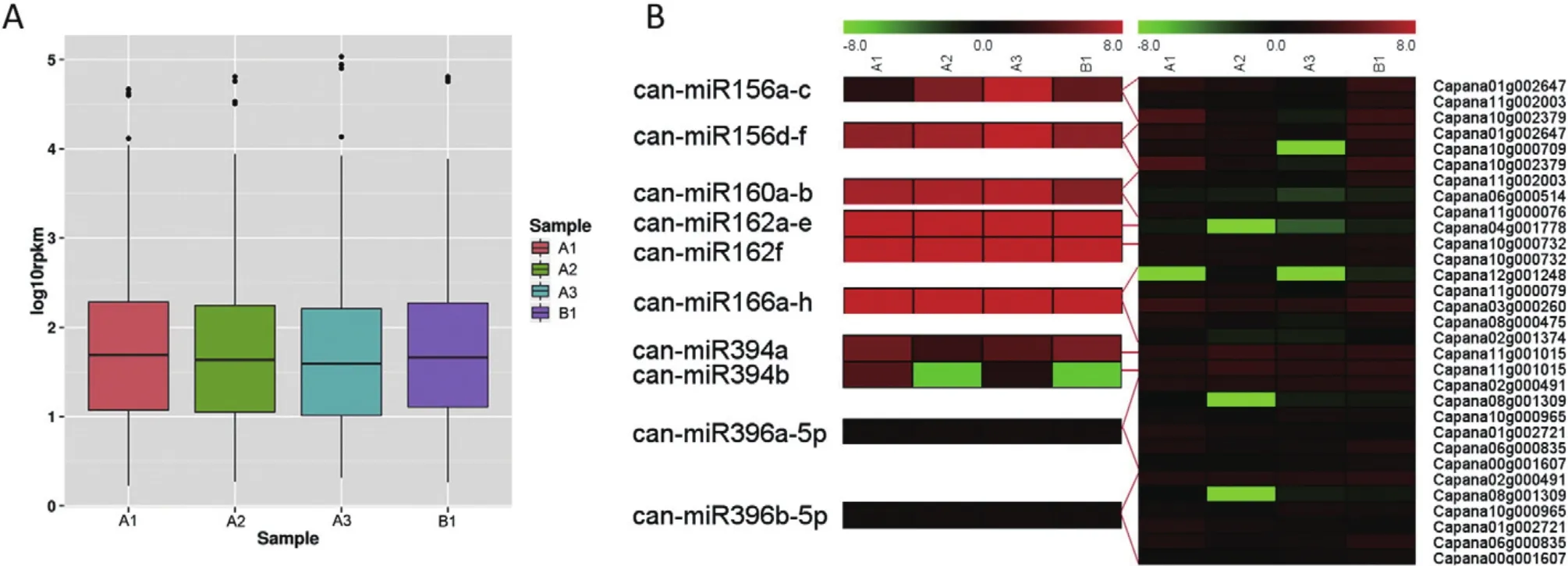
图5 miRNA的表达分析
Liu[34]等研究表明,miR394在水稻中与乙烯相关。Song[35]等研究表明miR394在油菜 (Brassica napus)中超表达后,延迟了油菜开花时间,且种荚和种子的体积均有增大。本研究中,深度测序和qRT-PCR的结果表显示can-miR394a在DAA50时期(A3)下调表达。表明can-miR394a可能与果实发育相关。
辣椒果实品质和风味很大程度上受到糖酸成分和含量的影响,因此糖酸代谢是辣椒果实发育最为重要的特征之一[36]。KEGG通路分析展示了“Luosijiao”(A1) 和“06 J19–1–1–1–2”(B1)果实的糖酸通路,如“氨基糖与核糖代谢”、“淀粉糖与蔗糖代谢”、“嘌呤代谢”和“N-糖链生物合成”(表S8)等。通过对靶基因进行注释,验证了3个miRNA(Novel_miR65、ovel_miR75、ovel_miR92)的靶基因半乳糖醛酸转移酶(Capana10g000025)参与了“蔗糖代谢(表S6)。以上结果表明,这些miRNA及其靶基因可能在果实发育与品质形成过程中起到重要的调节作用,但是需要更多的实验来证实其功能和调节作用。
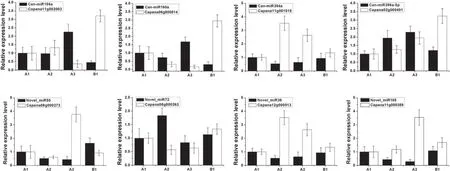
图6 qRT-PCR对部分miRNA及其靶基因的鉴定和表达分析
5 结论
本研究利用高通量测序技术鉴定了辣椒果实在不同发育时期的59个已知miRNA及310个新miRNA。通过差异表达分析揭示了这些miRNA在辣椒果实发育中的功能及作用。利用靶基因预测和GO富集分析,预测出310个新miRNA的靶基因,共656个。分析了一些果实发育和品质形成相关途径,如N-糖链生物合成、淀粉糖和蔗糖代谢、激素信号。通过生物信息学和实验相结合的手段,该研究为进一步研究辣椒及其他植物中microRNA在果实发育中的作用做了重要铺垫。
补充数据及相关文章可在线获取:http://dx.doi.org/10.1016/j.gene.2017.01.020。
致谢
本研究由中国农业研究系统专项基金(CARS-25-A-8)资助。
[1]Govindarajan V S. Capsicum production, technology,chemistry and quality. Part 1: History, botany, cultivation and primary processing[J]. Crit Rev Food Sci, 1985,22:109-176
[2]Aza-González C, Nún~ez-Palenius H G, Ochoa-Alejo N.Molecular biology of capsaicinoid biosynthesis in chili pepper (Capsicum spp.) [J]. Plant Cell Rep, 2011, 30:695-706
[3]Eggink P M, Tikunov Y, Maliepaard C, Haanstra J P W, de Rooij H, Vogelaar A, Gutteling E W, Freymark G, Bovy A G, Visser R G F. Capturing flavors from Capsicum baccatum by introgression in sweet pepper[J]. Theor Appl Genet, 2014, 127:373-390
[4]do Rêgo E R, do Rêgo M M, Cruz C D, Finger F L, Casali V W D. Phenotypic diversity, correlation and importance of variables for fruit quality and yield traits in Brazilian peppers (Capsicum baccatum) [J]. Genet Resour Crop Evol, 2011, 58:909-918
[5]Isabelle M, Lee B L, Lim M T, Koh W-P, Huang D, Ong C N. Antioxidant activity and pro fi les of common fruits in Singapore[J]. Food Chem, 2010, 123:77-84
[6]Perla V, Nimmakayala P, Nadimi M, Alaparthi S, Hankins G R, Ebert A W, Reddy U K. Vitamin C and reducing sugars in the world collection of Capsicum baccatum L.genotypes[J]. Food Chem, 2016, 202:189-198
[7]Martínez-López L A, Ochoa-Alejo N, Martínez O.Dynamics of the chili pepper transcriptome during fruit development[J]. BMC Genomics, 2014, 15: 143
[8]Liu S, Li W, Wu Y, Chen C, Lei J. De novo transcriptome assembly in chili pepper (Capsicum frutescens) to identify genes involved in the biosynthesis of capsaicinoids[J].PLoS One, 2013, 8, e48156
[9]Hu J, Zhang H, Ding Y. Identification of conserved microRNAs and their targets in the model legume Lotus japonicus[J]. J. Biotechnol, 2013, 164:520-524
[10]Chen X. Small RNAs and their roles in plant development[J]. Annu. Rev. Cell Dev. Biol., 2009, 25:21-44
[11]Bartel D P. MicroRNAs: Genomics, biogenesis,mechanism and function[J]. Cell, 2004, 116:281-297
[12]Qin C, Yu C, Shen Y, Fang X, Chen L, Min J, Cheng J, Zhao S, Xu M, Luo Y, Yang Y, Wu Z, Mao L, Wu H,Ling-Hu C, Zhou H, Lin H, Gonzalez-Morales S, Trejo-Saavedra D L, Tian H, et al. Whole-genome sequencing of cultivated and wild peppers provides insights into Capsicum domestication and specialization[J]. Proc Natl.Acad. Sci., 2014, 111:5135-5140
[13]Kim S, Park M, Yeom S, Kim Y, Lee J M, Lee H, Seo E, Choi J, Cheong K, Kim K, Jung K, Lee G, Oh S, Bae C, Kim S, Lee H, Kim S, Kim M, Kang B, Jo Y D, et al.Genome sequence of the hot pepper provides insights into the evolution of pungency in Capsicum species[J]. Nat Genet, 2014, 46:270
[14]Hwang D G, Park J H, Lim J Y, Kim D, Choi Y, Kim S, Reeves G, Yeom S I, Lee J S, Park M, Kim S, Choi I Y, Choi D, Shin C. The hot pepper (Capsicum annuum)microRNA transcriptome reveals novel and conserved targets: a foundation for understanding microRNA functional roles in hot pepper[J]. PLoS One, 2013, 8,e64238
[15]Friedlaender M R, Mackowiak S D, Li N, Chen W,Rajewsky N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades[J]. Nucleic Acids Res, 2012, 40:37-52
[16]Meyers B C, Axtell M J, Bartel B, Bartel D P, Baulcombe D, Bowman J L, Cao X F, Carrington J C, Chen X M,Green P J, Griffiths-Jones S, Jacobsen S E, Mallory A C, Martienssen R A, Poethig R S, Qi Y J, Vaucheret H,Voinnet O, Watanabe Y, Weigel D, zhu J K. Criteria for annotation of plant MicroRNAs[J]. Plant Cell, 2008,20:3186-3190
[17]Wang L, Feng Z, Wang X, Wang X W, Zhang X G.DEGseq: an R package for identifying differentially expressed genes from RNA-seq data[J]. Bioinformatics,2010, 26:136-138
[18]Dai X, Zhao P X. psRNATarget: a plant small RNA target analysis server[J]. Nucleic Acids Res, 2011,392:W155-W159
[19]Alexa A, Rahnenfuehrer J, Lengauer T. Improved scoring of functional groups from gene expression data by decorrelating GO graph structure[J]. Bioinformatics, 2006,22:1600-1607
[20]Varkonyi-Gasic E, Wu R, Wood M, Walton E F, Hellens R P. Protocol: a highly sensitive RT-PCR method for detection and quantification of microRNAs[J]. Plant Methods, 2007, 3: 12
[21]Livak K J, Schmittgen T D. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCTmethod[J]. Methods, 2001, 25:402-408
[22]Kudla G, Lipinski L, Caf fi n F, Helwak A, Zylicz M. High guanine and cytosine content increases mRNA levels in mammalian cells[J]. PLoS Biol, 2006, 4:933-942
[23]Zhang X H, Zou Z, Zhang J H, Zhang Y Y, Han Q Q, Hu T X, Xu X G, Liu H, Li H X, Ye Z B. Over-expression of sly-miR156a in tomato results in multiple vegetative and reproductive trait alterations and partial phenocopy of the sft mutant[J]. FEBS Lett, 2011, 585:435-439
[24]Zuker M. Mfold web server for nucleic acid folding and hybridization prediction[J]. Nucleic Acids Res, 2003,31:3406-3415
[25]Jones-Rhoades M W, Bartel D P, Bartel B. MicroRNAs and their regulatory roles in plants[J]. Annu. Rev. Plant Biol., 2006, 57:19-53
[26]Hu J, Sun L, Ding Y. Identification of conserved microRNAs and their targets in chickpea (Cicer arietinum L.) [J]. Plant Signaling & Behavior, 2013, 8:e23604
[27]Wu G, Poethig R S. Temporal regulation of shoot development in Arabidopsis thaliana by miR156 and its target SPL3[J]. Development, 2006, 133:3539-3547
[28]Bai S L, Saito T, Ito A, Tuan P A, Xu Y, Teng Y W,Moriguchi T. Small RNA and PARE sequencing in fl ower bud reveal the involvement of sRNAs in endodormancy release of Japanese pear (Pyrus pyrifolia 'Kosui') [J]. BMC Genomics, 2016, 17: 230
[29]Ferreira e Silva G F, Silva E M, Azevedo M D S, Guivin M A C, Ramiro D A, Figueiredo C R, Carrer H, Peres L E P, Nogueira F T S. microRNA156-targeted SPL/SBP box transcription factors regulate tomato ovary and fruit development[J]. The Plant journal: for cell and molecular biology, 2014, 78:604-618
[30]Sun C, Zhao Q, Liu D D, You C X, Hao Y J. Ectopic expression of the apple Md-miRNA156h gene regulates fl ower and fruit development in Arabidopsis[J]. Plant Cell Tiss Organ Cult, 2013, 112:343-351
[31]Mallory A C, Bartel D P, Bartel B. MicroRNA-directed regulation of Arabidopsis AUXIN RESPONSE FACTOR17 is essential for proper development and modulates expression of early auxin response genes[J]. Plant Cell,2005, 17:1360-1375
[32]Wu M F, Tian Q, Reed J W. Arabidopsis microRNA167 controls patterns of ARF6 and ARF8 expression,and regulates both female and male reproduction[J].Development, 2006, 133:4211-4218
[33]Wang X, Kong H, Ma H. F-box proteins regulate ethylene signaling and more[J]. Gene Dev, 2009, 23:391-396
[34]Liu Q, Zhang Y C, Wang C Y, Luo Y C, Huang Q J, Chen S Y, Zhou H, Qu L H, Chen Y Q. Expression analysis of phytohormone-regulated microRNAs in rice, implying their regulation roles in plant hormone signaling[J]. FEBS Lett, 2009, 583:723-728
[35]Song J B, Shu X X, Shen Q, Li B W, Song J, Yang Z M.Altered fruit and seed development of transgenic rapeseed(Brassica napus) over-expressing MicroRNA394[J]. PLoS One, 2015, 10, e0125427
[36]Wu J, Wang D F, Liu Y F, Wang L, Qiao X, Zhang S L. Identification of miRNAs involved in pear fruit development and quality[J]. BMC Genomics, 2014,15:953-972
文献来源: Gene, 2017, 608: 66-72
缩略词DAA,花后天数;RISC,RNA诱导的沉默复合体;KOG,同源蛋白质簇;KEGG,京都基因及基因组百科全书;GO,基因本体论;AGO,Agronaute蛋白。
*通信作者:ZHANG Zhuqing , MA Yanqing , ZOU Xuexiao
**通信地址:ZOU Xuexiao,湖南大学研究生隆平分院,湖南省蔬菜研究所,长沙410125
电子邮件:cszzq@126.com (ZHANG Zhuqing);yanqingmahn@163.com (MA Yanqing);zouxuexiao428@163.com (ZOU Xuexiao)
猜你喜欢
新民周刊(2022年27期)2022-08-01 07:04:49
少儿科学周刊·少年版(2022年18期)2022-05-30 10:48:04
少儿科学周刊·少年版(2022年18期)2022-05-30 10:48:04
小哥白尼(神奇星球)(2021年6期)2021-07-28 06:31:36
传染病信息(2021年6期)2021-02-12 01:52:58
中外文摘(2020年9期)2020-06-01 13:47:56
中华家教(2018年7期)2018-08-01 06:32:38
文学少年(原创儿童文学)(2016年16期)2016-02-28 17:50:19
中国当代医药(2015年7期)2015-03-01 02:01:09
生物医学工程学进展(2015年1期)2015-02-28 14:53:42
