
UPLC-MS法检测动物类药材中氯霉素类药物残留Δ
2017-11-09 08:33:32张治军周文杰李志芸广西壮族自治区桂林食品药品检验所广西桂林5400桂林医学院药学院广西桂林54004
中国药房 2017年30期
张治军,周文杰,李志芸(.广西壮族自治区桂林食品药品检验所,广西桂林5400;.桂林医学院药学院,广西桂林54004)
UPLC-MS法检测动物类药材中氯霉素类药物残留Δ
张治军1*,周文杰1,李志芸2(1.广西壮族自治区桂林食品药品检验所,广西桂林541001;2.桂林医学院药学院,广西桂林541004)
目的:建立检测动物类药材中氯霉素类药物残留的方法。方法:采用超高效液相色谱-质谱法。色谱柱为SB-C18,流动相为乙腈-水(梯度洗脱),流速为0.2 mL/min,柱温为35℃,进样量为20 μL;离子源为电喷雾离子源,干燥气温度为350℃,干燥气流量为5 L/min,鞘气温度为250℃,鞘气流量为11 L/min,毛细管电压为3 500 V,负离子扫描方式,多反应检测模式。结果:氯霉素、氟甲砜霉素和甲砜霉素检测质量浓度线性范围均为0.5~15 ng/mL(r分别为0.999 8、0.999 9、0.998 9);定量限分别为0.03、0.03、0.15 μg/kg,检测限分别为0.01、0.01、0.05 μg/kg;精密度、稳定性、重复性试验的RSD<3.0%;回收率分别为84.00%~112.80%(RSD=10.15%,n=9)、88.24%~109.80%(RSD=7.11%,n=9)、88.24%~99.02%(RSD=3.91%,n=9)。结论:该方法操作简便,精密度、稳定性、重复性好,可用于动物类药材中氯霉素类药物残留的检测。
动物类药材;氯霉素类药物残留;超高效液相色谱-质谱法
动物类药材的应用在我国有着悠久历史,在中药材的发展史中有着重要的地位。随着自然资源的匮乏,不少动物类药材来源由野生变为人工养殖。但在养殖过程中,有些不法商家为了追求不正当利益,非法过量使用抗生素、雌激素等化学药物,从而造成相关药物残留,最终可能对人体造成损害。而氯霉素类药物因具有广谱抗菌能力,价格便宜,抑菌效果好,常被广泛用于养殖业。这类药物性质稳定、不易降解,极易造成残留。已有研究证明,长期摄入氯霉素类药物会严重损伤人体造血机能,可引起粒细胞缺乏症、再生障碍性贫血,同时对消化系统也具有较严重的毒副作用[1-3]。因此,欧盟国家、美国、我国及其他一些国家严禁该类药物在食用动物上使用,我国农业部235号公告(2002年)中明确把氯霉素列为食品动物养殖中的禁用药物[4]。但是对于动物类药材,有关氯霉素类药物残留的检测研究目前尚未见文献报道。本试验以常用动物类药材鸡内金、土鳖虫、地龙、水蛭为例,建立了以超高效液相色谱-质谱法(UPLC-MS)同时测定其中氯霉素、氟甲砜霉素和甲砜霉素残留的方法,并对市售120批动物类药材进行了检测,旨在为动物类药材中氯霉素类药物残留的检测和控制提出参考。
1 材料
1.1 仪器
1290Infinity型UPLC仪和6460型三重四极杆MS仪(美国Agilent公司);XS205DU型电子分析天平(瑞士Mettler-Toledo公司);3-30K型高速冷冻离心机(美国Sigma公司)。
1.2 试剂
氯霉素对照品(中国食品药品检定研究院,批号:130555-201203,纯度:99.80%);氟甲砜霉素对照品(批号:01103,纯度:99.0%)、甲砜霉素对照品(批号:01103,纯度:98.5%)、D5-氯霉素对照品(批号:10307AC,纯度:99.0%)均购于德国Dr.Ehrenstorfer GmbH公司;甲醇、乙腈、乙酸乙酯、正己烷均为色谱纯,其余试剂均为分析纯,水为纯化水。
1.3 药材
地龙、水蛭、鸡内金、土鳖虫药材各30批,均购于广西不同地区药店和药材市场,经广西桂林食品药品检验所饶伟文主任药师鉴定为真品。
2 方法与结果
2.1 试验条件
2.1.1 色谱条件色谱柱:SB-C1(850 mm×2.1 mm,1.8 μm);流动相:乙腈(A)-水(B),梯度洗脱(0~3 min,25%→70%A;3~5 min,70%→95%A;5~8 min,95%→25%A);流速:0.2 mL/min;柱温:35℃;进样量:20 μL。
2.1.2 质谱条件离子源:电喷雾离子源(ESI);干燥气温度:350℃;干燥气流量:5 L/min;鞘气温度:250℃;鞘气流量:11 L/min;毛细管电压:3 500 V;负离子扫描方式,多反应检测模式。质谱分析参数见表1。
2.2 溶液的制备
2.2.1 混合对照品溶液精密称取待测成分对照品各10.0 mg,置于20 mL量瓶中,加D5-氯霉素内标溶液(10 ng/mL)0.10 mL,加乙腈溶解并定容,摇匀,制成氯霉素、氟甲砜霉素、甲砜霉素质量浓度均为500 μg/mL的混合对照品溶液。
2.2.2 供试品溶液取药材样品2.0 g,置于50 mL离心管中,加D5-氯霉素内标溶液(10 ng/mL)0.10 mL,加碱化乙酸乙酯溶液(乙酸乙酯-25%氨水,97∶3,V/V,下同)25 mL,涡旋后水平振荡5 min,以半径12 cm、5 000 r/min离心10 min,取上清液,于45水浴氮气吹干;残渣加碱化乙酸乙酯溶液25 mL,涡旋后水平振荡5 min,以半径12 cm、5 000 r/min离心10 min,取上清液;合并上述上清液,于45℃水浴氮气吹干,残渣加4%氯化钠溶液5 mL溶解,加正己烷萃取2次,每次5 mL,弃去正己烷,再加乙酸乙酯萃取2次,每次10 mL,合并乙酸乙酯萃取液,于45℃水浴氮气吹干,残渣加1 mL水溶解,经0.22 μm微孔滤膜滤过,取续滤液,即得。
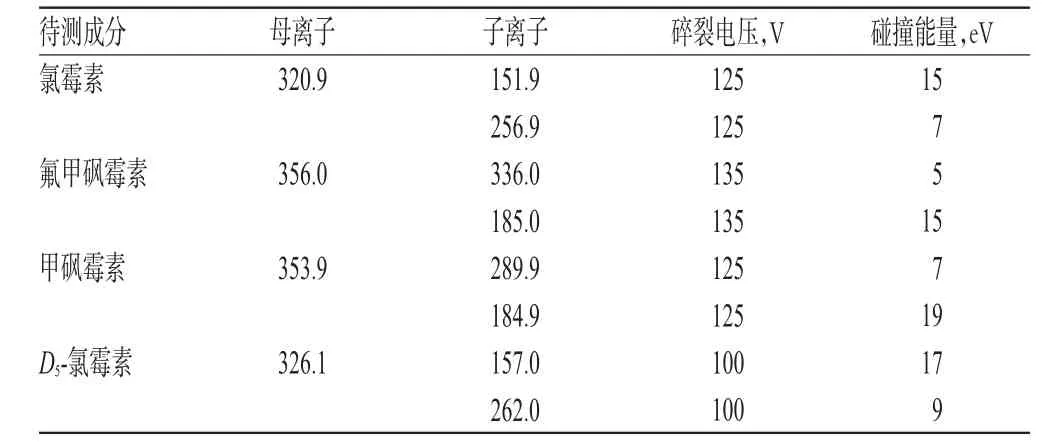
表1 质谱分析参数Tab 1 Mass spectrometry analysis conditions
2.3 系统适用性试验
取“2.2.1”项下混合对照品溶液适量,按“2.1”项下试验条件进样测定,记录色谱,详见图1。结果,氯霉素、氟甲砜霉素与甲砜霉素色谱峰可达到很好的分离,℃且无杂质峰干扰。
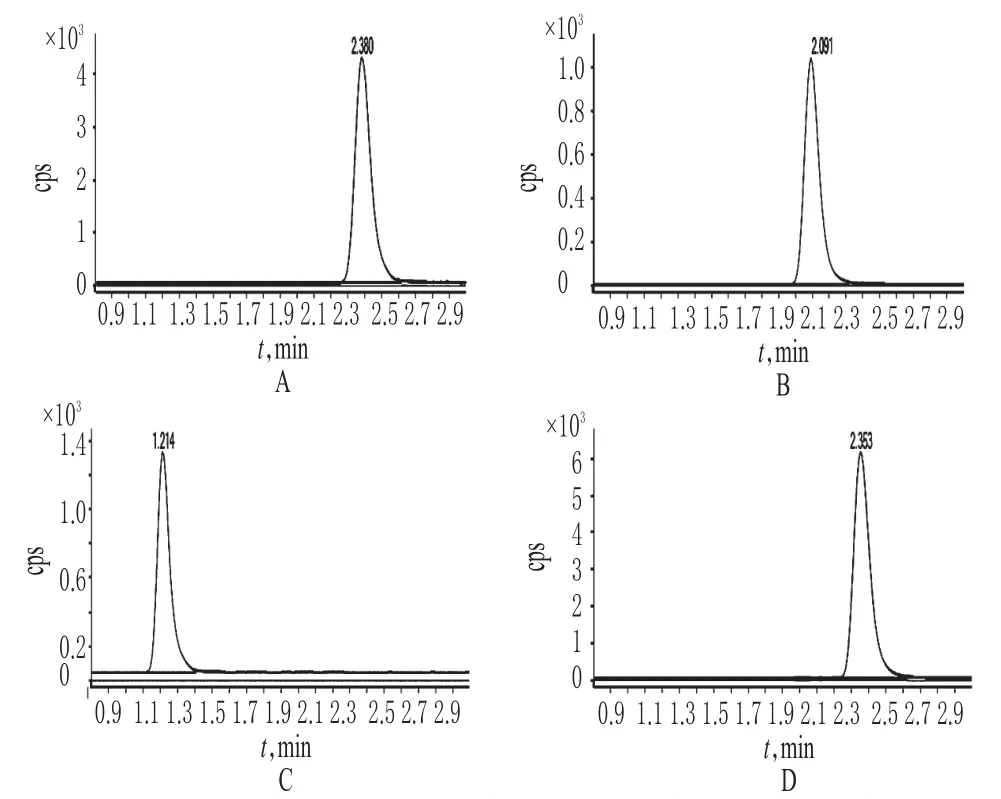
图1 提取离子流图Fig 1 Extraction particle flow graph
2.4 线性关系考察
分别精密量取“2.2.1”项下混合对照品溶液适量,加乙腈制成氯霉素、氟甲砜霉素、甲砜霉素质量浓度均为0.5、1.0、2.0、5.0、10.0、15.0 ng/mL的系列混合对照品溶液(均含D5-氯霉素内标溶液10 ng/mL)。精密量取上述系列混合对照品溶液各20 μL,按“2.1”项下试验条件进样测定,记录峰面积。以氯霉素、氟甲砜霉素、甲砜霉素质量浓度(x,ng/mL)为横坐标、峰面积(y)为纵坐标进行线性回归,得氯霉素、氟甲砜霉素、甲砜霉素回归方程分别为y=1.909 674x-0.000 299 161(r=0.999 8)、y=3.926 487x+0.007 515(r=0.999 9)、y=0.658 704x+0.004 180(r=0.998 9)。结果表明,氯霉素、氟甲砜霉素、甲砜霉素检测质量浓度线性范围均为0.5~15 ng/mL。
2.5 定量限与检测限考察
精密量取“2.2.1”项下混合对照品溶液适量,倍比稀释,并按“2.1”项下试验条件进样测定,当信噪比为10∶1时,得定量限;当信噪比为3∶1时,得检测限。结果,氯霉素、氟甲砜霉素、甲砜霉素定量限分别为0.03、0.03、0.15 μg/kg,检测限分别为0.01、0.01、0.05 μg/kg。
2.6 精密度试验
取“2.2.1”项下混合对照品溶液适量,按“2.1”项下试验条件连续进样测定6次,记录峰面积。结果,氯霉素、氟甲砜霉素、甲砜霉素峰面积的RSD分别为1.4%、1.5%、2.6%(n=6),表明仪器精密度良好。
2.7 稳定性试验
取“2.2.2”项下土鳖虫药材供试品溶液适量,分别于室温下放置0、2、8、12、24 h时按“2.1”项下试验条件进样测定,记录峰面积。结果,氯霉素、氟甲砜霉素、甲砜霉素峰面积的RSD分别为1.6%、1.3%、1.5%(n=5),表明供试品溶液室温放置24 h内基本稳定。
2.8 重复性试验
精密称取同一批土鳖虫药材样品适量,按“2.2.2”项下方法制备供试品溶液,共6份,再按“2.1”项下试验条件进样测定,记录峰面积。结果,氯霉素、氟甲砜霉素、甲砜霉素峰面积的RSD分别为2.4%、1.8%、2.2%(n=6),表明本方法重复性良好。
2.9 回收率试验
分别取低、中、高质量的待测成分对照品,按“2.2.2”项下方法制备供试品溶液,再按“2.1”项下试验条件进样测定,记录峰面积并计算加样回收率,结果见表2。
2.10 药材样品中氯霉素类药物残留量检测
取120批药材样品各适量,分别按“2.2.2”项下方法制备供试品溶液,再按“2.1”项下试验条件进样测定,记录峰面积并计算样品含量,结果见表3(表中仅列检出氯霉素类药物残留的药材样品)。
由表3可知,检出氯霉素类药物残留的鸡内金药材有18批(检出率为60%)、地龙药材7批(检出率为23%)、土鳖虫药材4批(检出率为13%)、水蛭药材4批(检出率为13%)。
3 讨论
由于动物养殖者一味地追求经济利益,氯霉素类药物有意无意滥用现象在当前养殖业中普遍存在。同时,环境水质、土壤的污染也是造成该类药物残留的一个重要原因[5-8]。这两种原因极易导致动物类药材中该类药物残留,甚至在含动物类药材较多的制剂中也会有氯霉素类药物残留[9]。从本试验收集的药材样品检测结果可知,动物类药材中存在氯霉素类药物残留的现象比较普遍,其中鸡内金药材的残留情况最严重。因此,建议有关监管部门给予足够重视。

表2 回收率试验结果(n=9)Tab 2 Results of recovery tests(n=9)
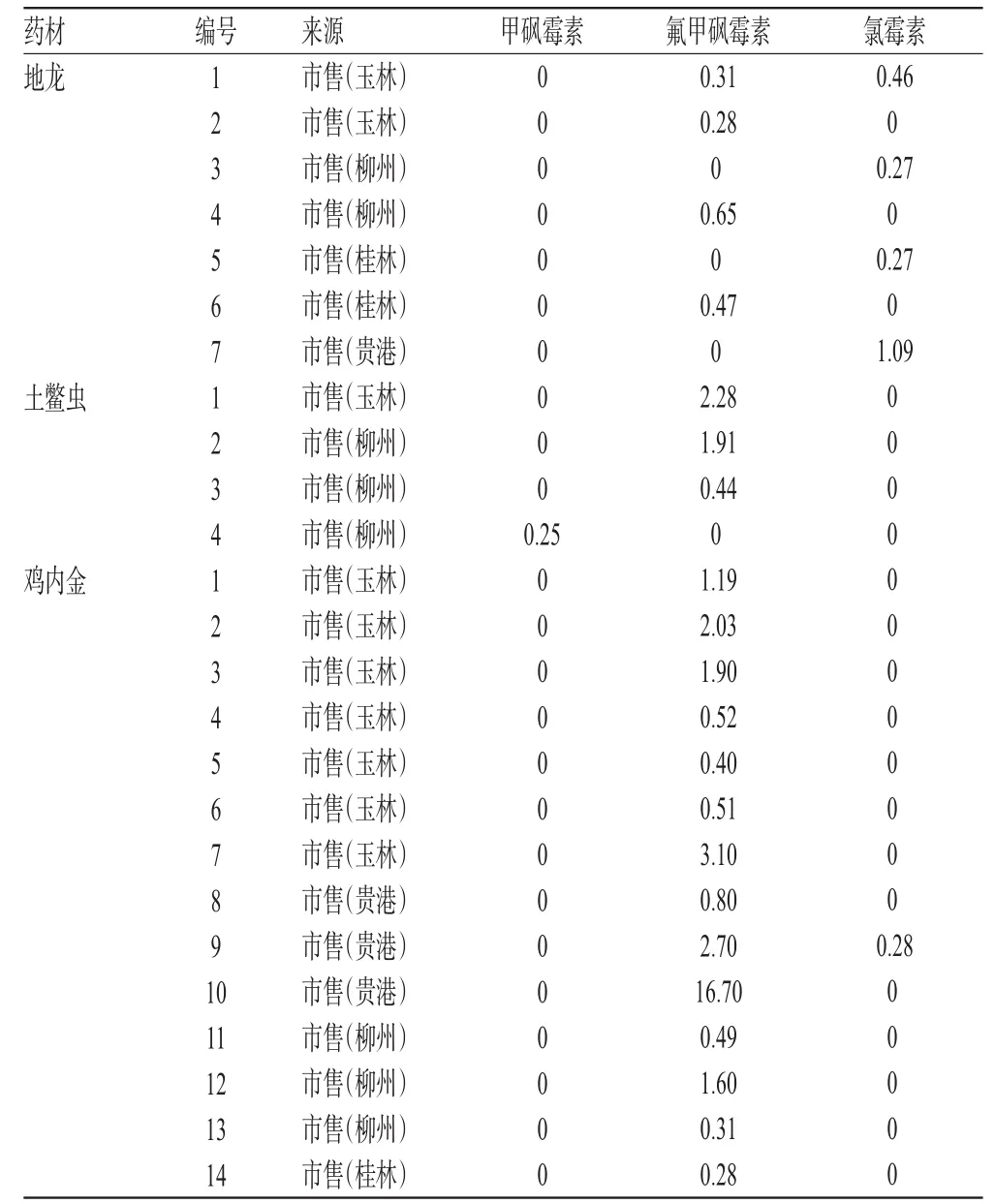
表3 药材样品中氯霉素类药物残留量检测结果(n=3,μg/kg)Tab 3 Results of chloramphenicol residue in medicinal herbs samples(n=3,μg/kg)
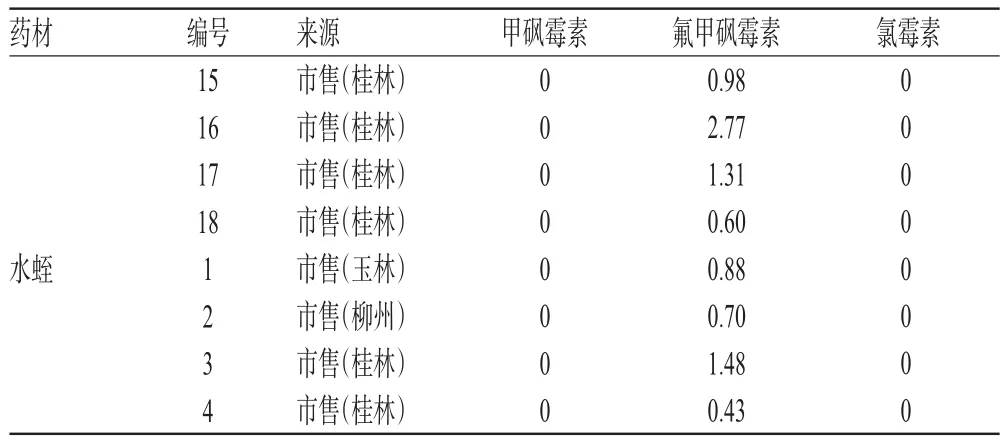
续表3Continued Tab 3
氯霉素类药物属于弱极性或弱碱性化合物,易溶于甲醇、乙腈、乙酸乙酯等有机溶剂。相关文献[10-15]报道的提取方法多采用乙腈或乙酸乙酯提取,并经固相萃取或正己烷液液萃取去除杂质。本试验曾比较了几种方法的提取效果,但结果要么基质效应很大,要么需要通过固相萃取柱净化,操作烦琐。经过进一步试验发现,采用乙酸乙酯加氨水提取药材样品,正己烷脱脂,乙酸乙酯萃取,操作简便,同时可消除药材样品的大部分基质效应,能得到较好的回收率和检测限。
综上所述,本方法操作简便,精密度、稳定性、重复性好,可用于动物类药材中氯霉素类药物残留的检测。
[1] 谭慧,麦琦.酶联免疫分析法测定水产品中氯霉素残留量[J].中国卫生检验杂志,2010,20(7):1649-1650.
[2] 胡红美,郭远明,雷科,等.分散固相萃取净化-气相色谱法测定水产品中氯霉素和氟苯尼考[J].食品科学,2014,35(8):231-235.
[3] 姚佳,王昕,张建新,等.免疫亲和柱-高效液相色谱法测定牛奶中氯霉素和玉米赤霉醇及其类似物[J].食品科学,2014,35(18):124-127.
[4] 农业部.关于发布《动物性食品中兽药最高残留限量》的通知:235号公告[S].2002.
[5] 王硕,张晶,邵兵.超高效液相色谱-串联质谱法测定污泥中氯霉素、磺胺、四环素与大环内脂类抗生素[J].分析测试学报,2013,32(3):179-185.
[6] 李红权,孙良娟,伍志强,等.高效液相色谱串联质谱法测定饲料中氯霉素、甲砜霉素、氟甲砜霉素残留[J].分析测试学报,2012,31(11):1396-1400.
[7] 华娟,方勤美,熊春娥,等.市售动物性食品中氯霉素类药物残留调查研究[J].食品安全质量检测学报,2013,4(1):165-170.
[8] 周亚民,叶领民.基于石英晶体微天平传感ELISA测定水体中残留氯霉素[J].东莞理工学院学报,2013,20(3):64-68.
[9] 冯向东,张玉洁,王戈,等.HLB固相萃取-HPLC-MS/MS法测定乌鸡白凤(小蜜丸)中氯霉素残留[J].中国药事,2016,30(10):1009-1014.
[10] 孙雷,张骊,王树槐,等.超高效液相色谱-串联质谱法检测动物源食品中氯霉素类药物及其代谢物残留[J].中国兽药杂志,2009,43(3):42-45.
[11] 李丽莉,罗轶.液相色谱-串联质谱法测定鸡肉中氯霉素残留[J].中国卫生检验杂志,2011,21(6):1357-1358.
[12] 曾玉梅,陈繁华.RP-HPLC法测定地砜软膏中3种主药含量[J].中国药房,2015,26(9):1271-1273.
[13] 胡音,洪建文.亚临界水萃取-HPLC法测定动物源食品中土霉素、四环素和氯霉素残留量[J].中国药房,2013,24(25):2356-2359.
[14] 陶昕晨,黄和,廖建萌,等.高效液相色谱-串联质谱法同时检测虾肉和猪肉中氯霉素、甲砜霉素、氟苯尼考和其代谢产物氟苯尼考胺残留[J].中国食品学报,2014,14(1):232-238.
[15] 胡争艳,吴平谷,王天娇,等.超高效液相色谱-串联质谱法同时测定鸡肉、鸡蛋中氯霉素和甲硝唑残留[J].中国卫生检验杂志,2016,26(21):3083-3097.
Determination of Chloramphenicol Residue in Animal Medicinal Herbs by UPLC-MS
ZHANG Zhijun1,ZHOU Wenjie1,LI Zhiyun2(1.Guangxi Guilin Institute for Food and Drug Control,Guangxi Guilin 541001,China;2.College of Pharmacy,Guilin Medical University,Guangxi Guilin 541004,China)
OBJECTIVE:To establish the method for determination of chloramphenicol residue in animal medicinal herbs.METHODS:UPLC-MS method was adopted.The determination was performed on SB-C18column with mobile phase consisted of acetonitrile-water(gradient elution)at the flow rate of 0.2 mL/min.The column temperature was 35℃,and sample size was 20 μL.The ion source was a jet stream ion focusing electrospray ion source.The temperature and flow of drying gas were 350℃and 5 L/min,and those of sheath gas were 250℃,11 L/min,and capillary voltage was 3 500 V.Negative ion scanning mode was conducted with multiple reaction monitoring(MRM)mode.RESULTS:The linear ranges of thiamphenicol,florfenicol and chloramphenicol were 0.5-15 ng/mL(r were 0.999 8,0.999 9,0.998 9,respectively).The limits of quantitation were 0.03,0.03,0.15 μg/kg,and the limits of detection were 0.01,0.01,0.05 μg/kg.RSDs of precision,stability and reproducibility tests were all lower than 3.0%.The recoveries were 84.00%-112.80%(RSD=10.15%,n=9),88.24%-109.80%(RSD=7.11%,n=9),88.24%-99.02%(RSD=3.91%,n=9)。CONCLUSIONS:The method is simple,precise,stable and reproducible,and can be used for determination of chloramphenicol in animal medicinal herbs.
Animal medicinal herbs;Chloramphenicol residue;UPLC-MS
R927
A
1001-0408(2017)30-4271-04
DOI10.6039/j.issn.1001-0408.2017.30.25
广西壮族自治区食品药品监督管理局科研项目(No.桂食药监科评函〔2015〕20号)
*副主任中药师。研究方向:药品质量检验、控制。E-mail:770013802@qq.com
2017-01-24
2017-04-06)
(编辑:张静)
猜你喜欢
小作家报·教研博览(2022年2期)2022-02-24 05:38:11
食品安全导刊(2021年20期)2021-08-30 06:40:34
中国民间疗法(2021年4期)2021-06-09 09:20:04
动漫界·幼教365(中班)(2021年4期)2021-05-23 20:32:55
北方文学·中旬(2016年4期)2016-06-08 09:07:10
家庭科学·新健康(2016年5期)2016-05-12 23:51:56
中国卫生标准管理(2015年25期)2016-01-14 09:29:24
湖南中医药大学学报(2015年1期)2016-01-06 01:06:36
中国药物应用与监测(2015年5期)2015-12-11 03:15:53
中国卫生标准管理(2015年15期)2015-01-26 20:32:38
