
高通量测序分析重度寻常痤疮患者和健康人肠道菌群差异
2017-11-02 05:51闫慧敏赵惠娟郭独一张春雷姜薇
中华皮肤科杂志 2017年3期
闫慧敏 赵惠娟 郭独一 张春雷 姜薇
100191北京大学第三医院皮肤科
高通量测序分析重度寻常痤疮患者和健康人肠道菌群差异
闫慧敏 赵惠娟 郭独一 张春雷 姜薇
100191北京大学第三医院皮肤科
目的利用高通量测序技术分析重度寻常痤疮患者和健康人肠道菌群的差异。方法门诊确诊的重度寻常痤疮患者10例及年龄、性别相匹配的健康人10例,分别收集粪便标本,提取细菌DNA,行16S rRNA测序鉴定微生物的种类,并进行菌群差异分析。结果两组肠道菌群多样性未发现显著差异,仅在肠道菌群的相对丰度上发现个别具有显著差异的菌。两组标本主要由拟杆菌门、厚壁菌门、变形菌门、放线菌门组成。比较两组在门、属水平上菌群相对丰度,差异均无统计学意义;在种水平上,Blautia producta、Coprococcus eutactus在两组间差异有统计学意义。结论重度寻常痤疮患者和健康人相比,菌群多样性指数未见明显差异。但某些细菌菌种存在显著差异。
寻常痤疮;肠道菌群;高通量测序
近期有学者提出,肠道微生物可能也参与寻常痤疮的发病[1],寻常痤疮患者发生胃肠道症状(口臭、胃反流、腹胀、便秘)的风险高于正常人[2]。我们选取10例重度寻常痤疮患者和10例年龄、性别相匹配的健康人,并收集他们的粪便,通过高通量测序细菌16S rRNA基因序列技术分析比较重度寻常痤疮患者与健康人肠道菌群分布的差异。
材料与方法
一、对象
2016年1-8月在北京大学第三医院皮肤科就诊的重度寻常痤疮(诊断标准参考中国痤疮治疗指南[3])患者(患者组)以及健康人群。重度寻常痤疮患者10例,男4例,女6例,年龄17~ 35(平均24.4)岁,体质指数(body mass index,BMI)<24。无皮肤病的健康对照组10例,男4例,女6例,年龄19~31(平均24.9)岁,BMI<24。两组入选者年龄、性别、体重差异均无统计学意义。所有研究对象的排除标准:过去2个月内局部或系统使用过抗生素、激素、维A酸类药物及免疫抑制剂;有胃肠道疾病及其他系统性疾病;孕期及哺乳期妇女。
二、试剂与方法
1.标本采集:收集上述20例研究对象的粪便于无菌容器中,-20℃储存。
2.DNA提取:采集标本后24 h内提取DNA,采用E.Z.N.A.®Stool DNA Kit DNA提取试剂盒(美国Omega公司)提取DNA,使用QubitFluorometer试剂盒(美国Life Technologies公司)检测DNA浓度,使用1%琼脂糖凝胶电泳检测样品完整性。DNA于-20℃储存。
3.生物信息学分析:提取患者组和对照组细菌总DNA后,送至深圳华大基因科技有限公司行16S rRNA基因V4区高通量测序,对测序结果进行生物信息学分析和数据分析。①使用FLASH软件将双末端测序得到的成对reads组装成1条序列,得到高变区的Tags;②用UPARSE软件以与16S rRNA序列相似度>97%对序列进行操作单元(OTU)聚类,得到OTU的代表序列,通过RDP classier软件将OTU代表序列与数据库(Greengene)比对进行物种注释,置信度阈值设置为0.8;③使用mothur软件(http://www.mothur.org/wiki/Calculators)分析微生物的群落多样性及丰度,Alpha多样性是对单个样品中物种多样性的分析,包括Observed species指数,Chao指数,Shannon指数,其中Observed species、Chao指数反映样品中群落的丰富度,Shannon指数反映群落的多样性,计算样本的Alpha多样性值并用R软件做出相应的稀释曲线图;④采用Beta多样性分析样品在物种多样性方面的差异,其中,Clustering聚类分析可以判断各样品的物种组成相似性,样品越靠近,枝长越短,说明两个样品的物种组成越相似;PCA分析是将多组数据的差异反映在二维坐标图上,两个样品距离越近,则表示两个样品的组成越相似。
三、统计学分析
采用Wilcoxon Rank-Sum Test分析两组样品在不同分类学水平的差异,以P≤0.05为差异有统计学意义。
结 果
一、样品测序与聚类
20份样品共测出641 745条高质量的近全长16S rRNA序列,平均每个样品32 087条,其中患者组共得到321 276条,对照组320 469条。20个样本中共聚类为588 OTUs,其中患者组467 OTUs,对照组501 OTUs,380 OTUs为两者共有。
二、Alpha多样性分析
Observed species指数稀释曲线、Chao指数稀释曲线均达到平台期,表示测序深度已基本覆盖到样品中所有的物种(图1、2);对照组和患者组的Shannon指数均分布在2.0~3.5之间(图3),两组的物种多样性未见明显差异。此外,Observed species指数分布、Chao指数分布、Shannon指数分布对照组与患者组差异均无统计学意义(P>0.05)。见表1。受样品群落中物种丰富度和物种均匀度的影响,相同物种丰富度情况下,群落中各物种具有越大的均匀度,则认为群落具有越大的多样性,由于所有样品的测序深度已达到要求,所以Shannon结果未受影响。
三、微生物分类
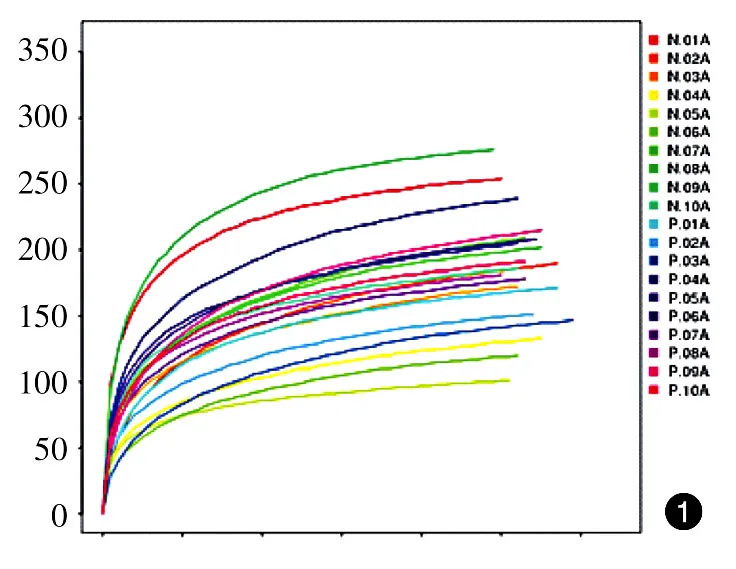
图1 10例重度寻常痤疮患者和10例健康对照粪便细菌总DNA Observed species指数稀释曲线
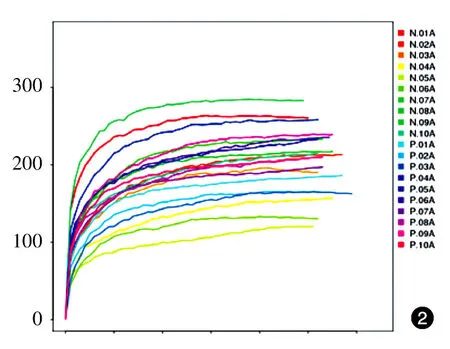
图2 10例重度寻常痤疮患者和10例健康对照粪便细菌总DNA Chao指数稀释曲线
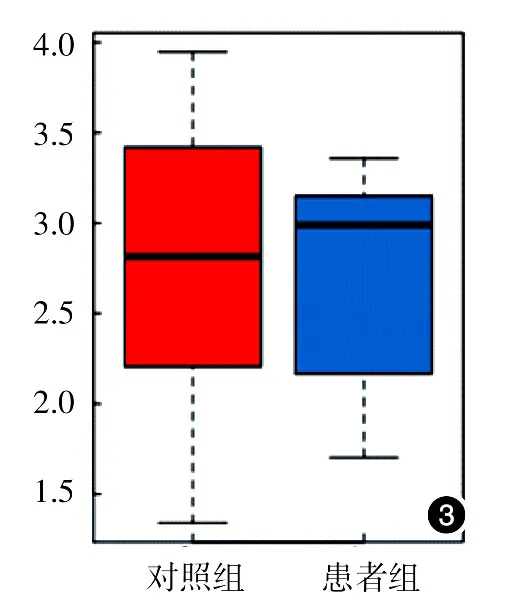
图3 10例重度寻常痤疮患者和10例健康对照粪便细菌总DNA Shannon指数盒形图
表1 重度寻常痤疮患者和健康对照肠道菌群样品Alpha多样性统计结果(±s)
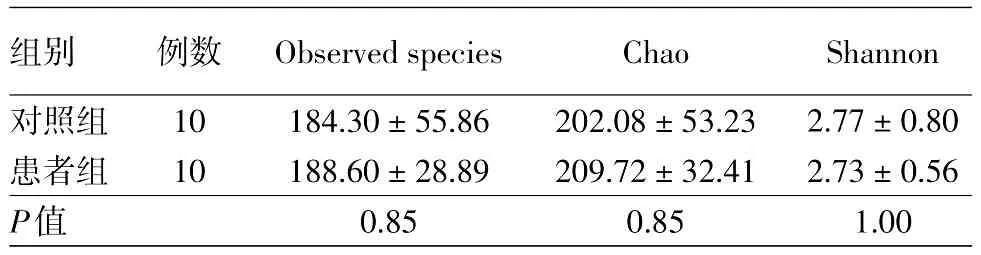
表1 重度寻常痤疮患者和健康对照肠道菌群样品Alpha多样性统计结果(±s)
注:n=20。Observed species指数、Chao指数反映样品中群落的丰富度,Shannon指数反映群落的多样性
组别对照组患者组P值例数10 10 Observed species 184.30±55.86 188.60±28.89 0.85 Chao 202.08±53.23 209.72±32.41 0.85 Shannon 2.77±0.80 2.73±0.56 1.00
588 OTUs分属于12个细菌门,其中11个细菌门为两组共有,迷踪菌门(Elusimicrobia)只存在于对照组,对照组和患者组大部分的肠道菌群属于拟杆菌门(Bacteroidetes)、厚壁菌门(Firmcutes)、变形菌门(Proteobacteria)、放线菌门(Actinobacteria),见图4。对照组与患者组的平均丰度拟杆菌门最高分别为58.71%、60.80%,其次是厚壁菌门31.24%、30.59%,变形菌门6.64%、6.34%,放线菌门1.84%、0.35%,梭杆菌门(Fusobacteria)1.39%、1.82%,两组差异均无统计学意义(P>0.05)。其中,有1例患者和1例健康人梭杆菌门丰度较高。
分析属水平,20份标本共鉴定出83个细菌属,其中拟杆菌属(Bacteroides)、普氏菌属(Prevotella)、布劳特菌(Blautia)、梭杆菌属(Faecalibacterium)、梭菌属(Fusobacterium)、毛螺菌属(Lachnospira)、巨单胞菌属(Megamonas)、颤螺菌属(Oscillospira)、巨球菌属(Megasphaera)、Parabacteroides、萨特菌属(Sutterella)、罗氏菌属(Roseburia)、Phascolarctobacterium、瘤胃球菌属(Ruminococcus)、双歧杆菌属(Bifidobacterium)、鲸杆菌属(Cetobacterium)在两组中含量丰富(>1%)差异均无统计学意义(P>0.05)。见图5。
在种水平上,20份标本共鉴定出55个细菌种,在两组中丰度较高的种(>1%)有粪拟杆菌(Bacteroides caccae)、普通拟杆菌(Bacteroides plebeius)、单形拟杆菌(Bacteroides uniformis)、柔嫩梭菌群(Faecalibacterium prausnitzii)、普氏菌卡布里(Prevotella copri),其中,Blautia producta的平均丰度对照组为0.05%,患者组为0.02%,P=0.038;Coprococcus eutactus平均丰度对照组为0.18%,患者组为0.01%,差异均有统计学意义(P<0.05)。其他菌种组间差异均无统计学意义(P>0.05)。见图6。
四、Beta多样性分析
本研究中,Clustering聚类分析,对照组和患者组未聚集成两个明显的簇,差异无统计学意义(P>0.05);PCA分析,对照组和患者组样品彼此间的距离差异无统计学意义(P>0.05),两组微生物的种类及丰度均相似。见图7、8。
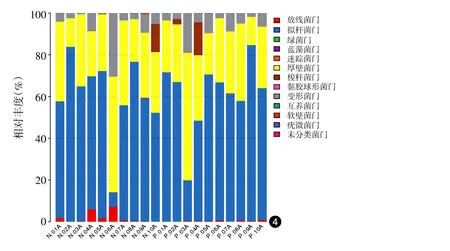
图4 10例重度寻常痤疮患者和10例健康对照粪便样品细菌门水平物种丰度图
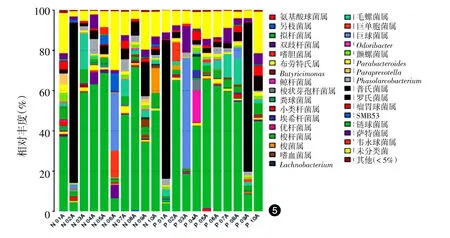
图5 10例重度寻常痤疮患者和10例健康对照粪便样品细菌属水平物种丰度图
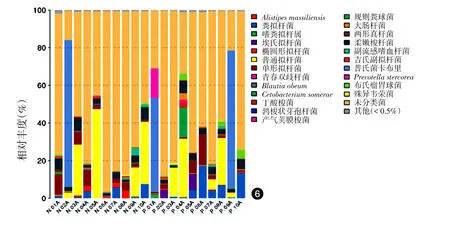
图6 10例重度寻常痤疮患者和10例健康对照粪便样品细菌种水平物种丰度图
讨 论
人类肠道系统大约有1 000多种细菌,在粪便中的微生物细胞密度约为1 013~1 014个/g,其中,70%的微生物位于结肠[4]。我们采用高通量测序方法分析重度寻常痤疮患者和健康人肠道微生物组成的差别。由于肥胖[5]、年龄[6]会影响肠道微生物的组成,故我们选取的研究对象为青年人,BMI< 24。
本研究共得到了641 745条序列,聚类成588 OTUs,分成12个门、83个属、55个种,两组常见的细菌门均是拟杆菌门、厚壁菌门、变形菌门,两组拟杆菌门、厚壁菌门之和占了肠道菌群的90%,拟杆菌门的含量更高,与其他两项国内研究的结果一致[6-7],但与美国人及埃及人的研究结果不同[8]。在美国人肠道研究显示,厚壁菌门含量最高,其次是拟杆菌门,且两者之比为1.4,埃及人为1.1,而本研究为0.5。这可能是由东西方饮食习惯不同造成的。有研究显示,高蛋白、高动物脂肪饮食会导致拟杆菌门含量高,而长期的碳水化合物饮食会导致厚壁菌门含量高[9],厚壁菌门/拟杆菌门的比值可作为潜在的人类肠道病理状况的的生物学指标[4]。本研究中两组厚壁菌门/拟杆菌门比值相同,说明患者组的肠道微生物和对照组相似,并非处于病理状况。
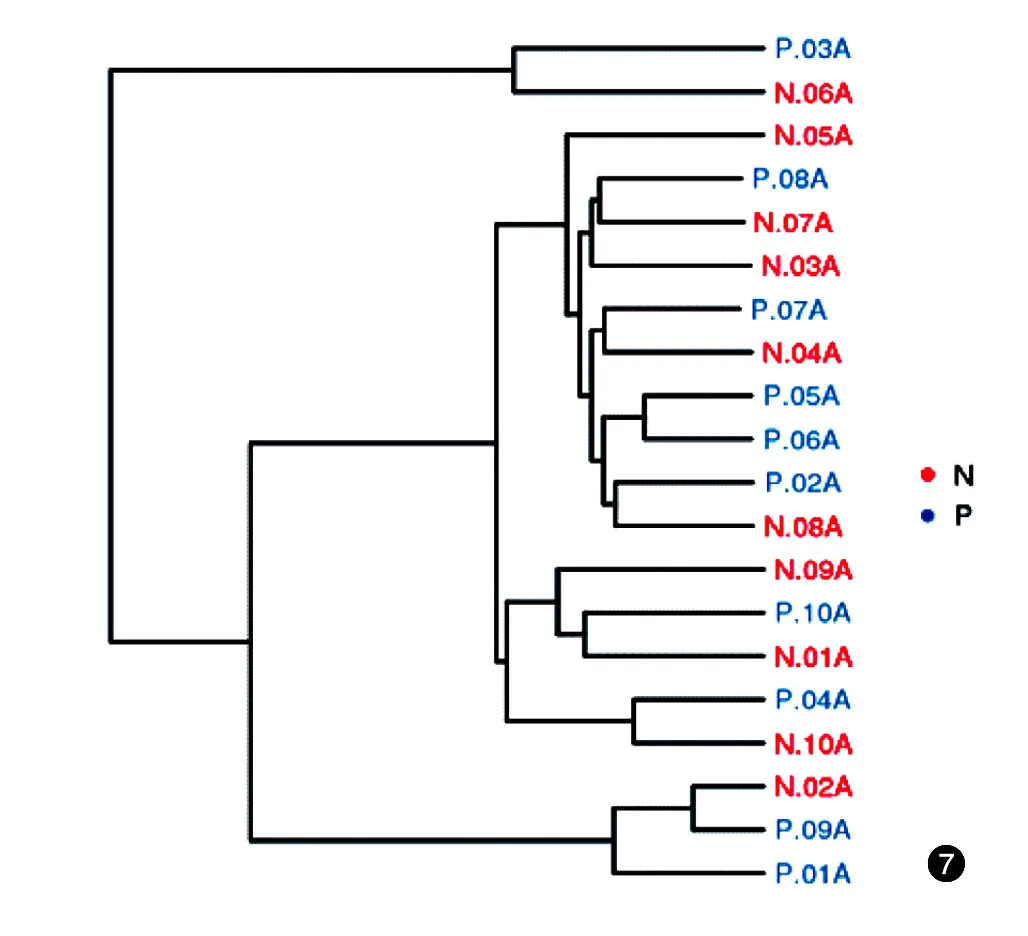
图7 10例重度寻常痤疮患者和10例健康对照粪便细菌总DNA clustering聚类图 N:健康对照组;P:患者组
Manzhalii等[10]发现,Bafidobacteria、Lactobacteria可以改善面部具有丘疹、脓疱患者的临床症状,而在本研究中,此两种菌在痤疮患者组的丰度与对照组无明显差异。Loveman等[11]为了验证肠道微生物是否参与寻常痤疮的发生,用培养的方法来分析寻常痤疮患者和正常人肠道微生物,未发现差异。由于传统的培养方法只能培养出肠道微生物的10%~30%[12],所以我们采用了现代分子生物学技术16S RNA测序来鉴定和分析肠道微生物菌群,结果发现Blautia producta、Coprococcus eutactus存在显著差异,寻常痤疮患者肠道内两种菌的比例明显降低。Blautia producta属于革兰染色阳性厌氧菌,厚壁菌门梭菌纲,它在肠道内与其他菌群一起加强肠道功能并抵抗病原菌的入侵[13],将不易消化的碳水化合物转化为短链脂肪酸为人体供能,还可与其他梭菌属一起增加肠道内丁酸盐的形成,并能降解黏蛋白、β 天冬氨酰甘氨酸、胆红素[14]。Coprococcus eutactus也属于梭菌纲,革兰阳性厌氧菌,也具有产生丁酸盐的能力[15]。丁酸盐主要在结肠形成,不仅具有维持肠道功能稳定的作用,且具有抗肿瘤、抗炎症、抗氧化的功能[16]。丁酸盐还可以调节结肠中调节性T细胞的活化和增殖[17],增加小鼠T细胞抑制效应性T细胞活化的能力[18]。有研究报道,肠易激综合征患者比健康人肠道内Coprococcus eutactus及其相关菌含量明显减少[19],轻型湿疹患者肠道内Coprococcus eutactus及其他产丁酸盐的细菌种类及丰度比重度湿疹患者增多,提示丁酸盐可减轻湿疹的症状[20]。这两种菌在寻常痤疮患者组肠道菌群中含量明显降低,肠道内丁酸盐形成减少,肠道稳态平衡被破坏,导致系统性炎症的发生,进而诱发或加重寻常痤疮的发生,但其具体机制仍需进一步的研究。
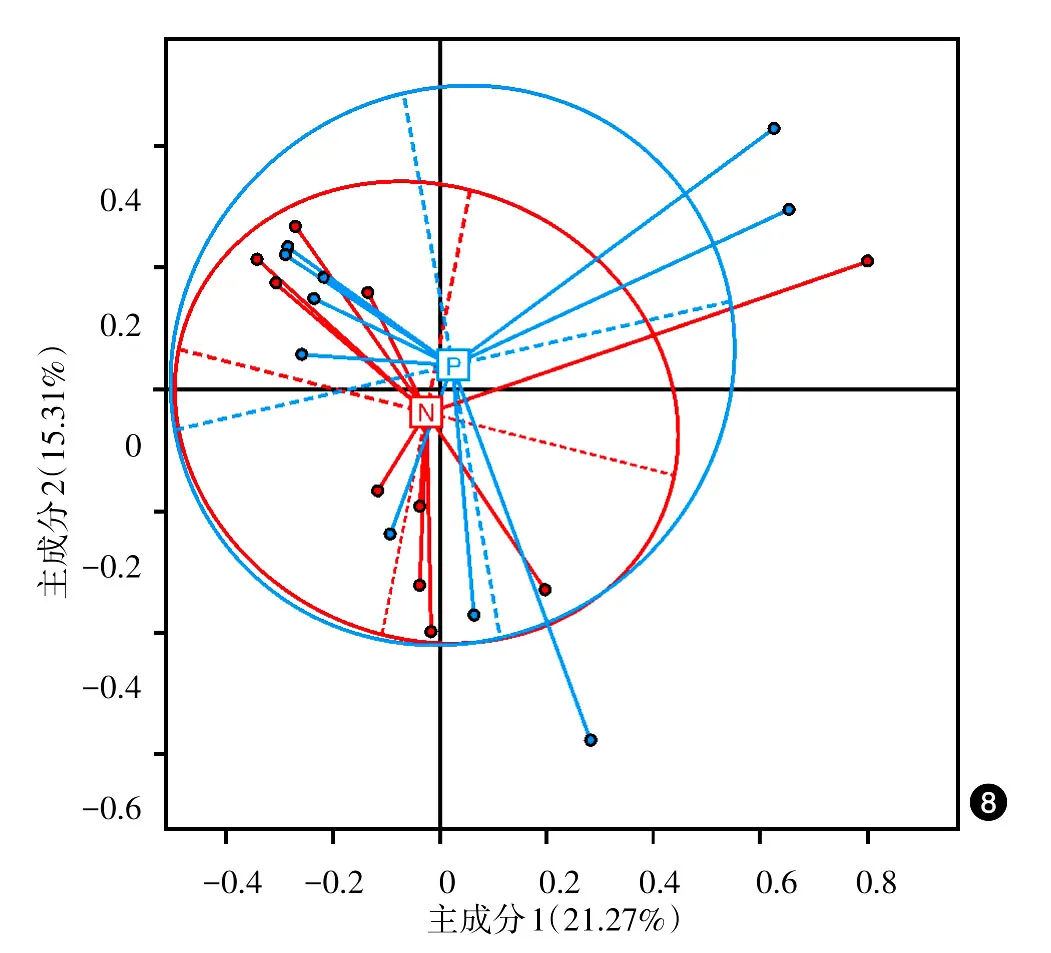
图8 10例重度寻常痤疮患者和10例健康对照粪便微生物群落主成分分析图 蓝色线条(P):重度寻常痤疮患者;红色线条(N):健康对照
本研究表明,寻常痤疮患者和健康人肠道菌群多样性无差异,在肠道微生物的相对丰度上发现个别具有显著差异的菌,即产丁酸盐的菌种(Blautia producta、Coprococcus eutactus),寻常痤疮患者组较健康组的产丁酸盐的菌种含量明显减少,提示产丁酸盐的相关菌可能具有减轻寻常痤疮症状的作用。
因本研究样本量相对较小,故未对性别进行分组,但性别对肠道微生物也有一定影响[21],16S rRNA序列分析技术对微生物的种类的鉴定仍存在一定局限性,因此肠道菌群对寻常痤疮发病影响的具体机制仍需进一步研究。
[1]Volkova LA,Khalif IL,Kabanova IN.Impact of the impaired intestinal microflora on the course of acne vulgaris[J].Klin Med(Mosk),2001,79(6):39-41.
[2]Zhang H,Liao W,Chao W,et al.Risk factors for sebaceous gland diseases and their relationship to gastrointestinal dysfunction in Han adolescents[J].J Dermatol,2008,35(9):555-561.DOI:10.1111/j.1346-8138.2008.00523.x.
[3]项蕾红.中国痤疮治疗指南(2014修订版)[J].临床皮肤科杂志,2015,44(1):52-57.DOI:10.16761/j.cnki.1000-4963.2015.01.020.
[4]Ley RE,Turnbaugh PJ,Klein S,et al.Microbial ecology:human gut microbes associated with obesity[J].Nature,2006,444(7122):1022-1023.DOI:10.1038/4441022a.
[5]Chiu CM,Huang WC,Weng SL,et al.Systematic analysis of the association between gut flora and obesity through high-throughput sequencing and bioinformatics approaches[J].Biomed Res Int,2014,2014:906168.DOI:10.1155/2014/906168.
[6]Wang F,Yu T,Huang G,et al.Gut microbiota community and its assembly associated with age and diet in Chinese centenarians[J].J Microbiol Biotechnol,2015,25(8):1195-1204.DOI:10.4014/jmb.1410.10014.
[7]Gu S,Chen Y,Zhang X,et al.Identification of key taxa that favor intestinal colonization of clostridium difficile in an adult Chinese population[J].Microbes Infect,2016,18(1):30-38.DOI:10.1016/j.micinf.2015.09.008.
[8]Aly AM,Adel A,El-Gendy AO,et al.Gut microbiome alterations in patients with stage 4 hepatitis C[J].Gut Pathog,2016,8(1):42.DOI:10.1186/s13099-016-0124-2.
[9]Wu GD,Chen J,Hoffmann C,et al.Linking long-term dietary patterns with gut microbial enterotypes[J].Science,2011,334(6052):105-108.DOI:10.1126/science.1208344.
[10]ManzhaliiE,HornussD,StremmelW.Intestinal-bornedermatoses significantly improved by oral application of Escherichia coli Nissle 1917[J].World J Gastroenterol,2016,22(23):5415-5421.DOI:10.3748/wjg.v22.i23.5415.
[11]Loveman DE,Noojin RO,Winkler CJ.Comparative studies of enteric bacterial flora in acne vulgaris[J].J Invest Dermatol,1955,25(3):135-137.DOI:10.1038/jid.1955.110.
[12]Sokol H,Seksik P.The intestinal microbiota in inflammatory bowel diseases:time to connect with the host[J].Curr Opin Gastroenterol,2010,26(4):327-331.DOI:10.1097/MOG.0b013e328339536b.
[13]Sharma R,Young C,Neu J.Molecular modulation of intestinal epithelial barrier:contribution of microbiota[J].J Biomed Biotechnol,2010,2010:305879.DOI:10.1155/2010/305879.
[14]Becker N,Kunath J,Loh G,et al.Human intestinal microbiota:characterization of a simplified and stable gnotobiotic rat model[J].Gut Microbes,2011,2(1):25-33.DOI:10.4161/gmic.2.1.14651.
[15]Zhang L,Mu C,He X,et al.Effects of dietary fibre source on microbiota composition in the large intestine of suckling piglets[J].FEMS MicrobiolLett,2016,363(14):fnw138.DOI:10.1093/femsle/fnw138.
[16]Klampfer L,Huang J,Sasazuki T,et al.Inhibition of interferon gamma signaling by the short chain fatty acid butyrate[J].Mol Cancer Res,2003,1(11):855-862.
[17]Furusawa Y,Obata Y,Fukuda S,et al.Commensal microbederived butyrate induces the differentiation of colonic regulatory T cells[J].Nature,2013,504(7480):446-450.DOI:10.1038/nature12721.
[18]Smith PM,Howitt MR,Panikov N,et al.The microbial metabolites,short-chain fatty acids,regulate colonic Treg cell homeostasis[J].Science,2013,341(6145):569-573.DOI:10.1126/science.1241165.
[19]Salonen A,de Vos WM,Palva A.Gastrointestinal microbiota in irritable bowel syndrome:present state and perspectives[J].Microbiology,2010,156(Pt 11):3205-3215.DOI:10.1099/mic.0.043257-0.
[20]Nylund L,Nermes M,Isolauri E,et al.Severity of atopic disease inversely correlates with intestinal microbiota diversity and butyrate-producing bacteria[J].Allergy,2015,70(2):241-244.DOI:10.1111/all.12549.
[21]Moreno-Indias I1,Sánchez-Alcoholado L1,Sánchez-Garrido MÁ,etal.Neonatalandrogen exposure causes persistentgut microbiota dysbiosis related to metabolic disease in adult female rats[J].Endocrinology,2016,157(12):4888-4898.DOI:10.1210/en.2016-1317.
High-throughput sequencing analysis of gut microbiome in patients with severe acne vulgaris and healthy individuals
Yan Huimin,Zhao Huijuan,Guo Duyi,Zhang Chunlei,Jiang Wei
Department of Dermatology,Peking University Third Hospital,Beijing 100191,China
Jiang Wei,Email:jiangwei7366@163.com
ObjectiveTo compare differences of gut microbiome between patients with severe acne vulgaris and healthy individuals by using high-throughput sequencing technology.MethodsStool samples were collected from 10 outpatients with severe acne vulgaris and 10 age-and sex-matched healthy controls.Then,the bacterial DNA was extracted and subjected to 16S rRNA sequencing for the identification of microbial species,and the differences of gut microbiome were compared between the patients and controls.ResultsThere were no significant differences in the diversity of intestinal microflora,but only the relative abundance of a few bacteria differed significantly between the two groups.Gut microbiome in the two groups mainly consisted ofBacteroidetes,Firmicutes,ProteobacteriaandActinobacteria.There were no significant differences in the relative abundance of bacteria at the phylum and genus levels between the two groups.However,the relative abundance ofBlautia productaandCoprococcus eutactusat the species level differed remarkably between the two groups.ConclusionNo significant differences in the bacterial diversity indices are found,but some bacterial species significantly differ between the patients with severe acne vulgaris and healthy controls.
Acne vulgaris;Gut microbiome;High-throughput sequencing
姜薇,Email:jiangwei7366@163.com
10.3760/cma.j.issn.0412-4030.2017.03.005
国家自然科学基金(81201216)
Fund program:National Natural Science Foundation of China(81201216)
2016-12-07)
(本文编辑:颜艳)
猜你喜欢
中国农学通报(2022年14期)2022-06-01
世界科学技术-中医药现代化(2022年2期)2022-05-25
油气田环境保护(2022年2期)2022-05-09
山东畜牧兽医(2021年5期)2021-06-07
中国沼气(2019年1期)2019-04-13
意林(2018年20期)2018-10-31
大自然探索(2017年6期)2017-06-27
婚育与健康(2016年6期)2016-05-14
创业家(2015年3期)2015-02-27
创业家(2015年2期)2015-02-27
