
黑曲霉葡萄糖氧化酶基因在毕赤酵母SMD1168中的表达
2017-11-01 08:12:39肖成建范新蕾李汉广余晓斌
食品与生物技术学报 2017年9期
陈 楠, 肖成建, 范新蕾, 李汉广, 余晓斌
(江南大学 生物工程学院,江苏 无锡 214122)
黑曲霉葡萄糖氧化酶基因在毕赤酵母SMD1168中的表达
陈 楠, 肖成建, 范新蕾, 李汉广, 余晓斌*
(江南大学 生物工程学院,江苏 无锡 214122)
在毕赤酵母SMD1168中利用乙醇氧化酶AOX1强启动子表达黑曲霉葡萄糖氧化酶(Glucose oxidase,GOD)。提取黑曲霉Aspergillus niger PCTC的基因组DNA,以此为模板进行PCR扩增获得葡萄糖氧化酶基因,将目的基因插入到具有AOX1强启动子的表达载体pPICZαA上,经电转化导入毕赤酵母SMD1168中。经zeocin抗性平板初筛、摇床复筛以及SDS-PAGE蛋白质电泳的检测,获得了一株产葡萄糖氧化酶活力的菌株,该株菌在30℃、200 r/min的培养条件下,经体积分数1.0%的甲醇诱导发酵1 d可获得1.12 U/mL的酶活。对该菌株进行了摇瓶产酶条件优化,其最佳发酵条件为:在pH 5、30℃下经体积分数1.5%甲醇诱导7 d,酶活为32 U/mL。
葡萄糖氧化酶;黑曲霉;毕赤酵母;异源表达;启动子
葡萄糖氧化酶(Glucose oxidase,GOD)系统命名为β-D-葡萄糖:氧化还原酶(EC1.1.3.4)。葡萄糖氧化酶能专一性地氧化β-D-葡萄糖,生成葡萄糖酸和过氧化氢,因而具有除氧、抗氧化、去除葡萄糖以及生成葡萄糖酸的作用,近几十年来被广泛应用于与人体健康密切相关的食品、医药、生物技术等行业[1]。例如,它能有效地改善小麦面筋的组织结构,提高面团的持气性、弹性、韧性和持水性,从而提升了面包的质量[2];在白葡萄酒生产中,加入GOD/CAT系统后,可有效防止酚类和多酚氧化酶对白葡萄酒产生的严重褐变[3];葡萄糖氧化酶催化产生的过氧化氢可在纺织品中充当漂白剂的作用[4];葡萄糖氧化酶也可作为抑制微生物的成分添加到口腔护理产品中[5];此外,葡萄糖氧化酶是新型传感器中的重要的组成部分,可以用来测定糖含量[6]。
目前,葡萄糖氧化酶广泛采用黑曲霉或青霉发酵进行大规模生产,但在此过程中常常伴有过氧化氢酶、淀粉酶及纤维素酶等大量杂蛋白的产生[7]。近10年来,巴斯德毕赤酵母表达系统的操作简单、表达量高、稳定性强、能进行多种翻译后加工修饰等特点使其成为目前应用最广泛的外源蛋白质的表达系统之一[8]。两种从青霉Penicillium variabile P16[9]和黑曲霉9029[10]获得的GOD基因都在巴斯德毕赤酵母中获得了表达,前者在发酵11 d的条件下酶活为0.33 U/mL,后者的酶活最高为1.62 U/mL。作者采用的黑曲霉PCTC与上述文献报道的霉菌、葡萄糖氧化酶基因序列的相似度分别为96%和99%。
作者从黑曲霉PCTC中克隆了GOD基因,并用其转化蛋白质缺陷型毕赤酵母SMD1168,以期获得高效表达GOD蛋白酶缺陷型毕赤酵母生产用菌株。
1 材料与方法
1.1 实验材料
1.1.1 菌株与质粒 黑曲霉菌株PCTC:由作者所在实验室保存,葡萄糖氧化酶基因来自该菌株;质粒pMD18-T:用来构建克隆载体;大肠杆菌JM109:用来构建和克隆目的基因;质粒pPICZαA:用来表达目的基因;毕赤酵母SMD1168:作为表达目的基因的宿主。
1.1.2 试剂与仪器 工具酶、PCR相关试剂、XhoI、NotI限制性内切酶、T4DNA连接酶:购自Takara公司;PmeI:购自纽英伦生物技术有限公司;真菌基因组DNA快速抽提试剂盒、质粒提取试剂盒、胶回收试剂盒、DNA标准相对分子质量marker和Amp(氨苄青霉素)等:购自上海生工生物有限公司;琼脂糖、辣根过氧化物酶、邻联茴香胺等:购自Sigma公司;丙烯酰胺、甲叉双丙烯酰胺:Bio-Rad产品;色谱纯甲醇及其他试剂(分析纯):均购自国药集团;PCR仪和电转化仪:Eppendorf公司;电泳仪:北京六一仪器厂。
1.1.3 培养基
1)Aspergillus niger PCTC活化培养基 (PDA培养基):马铃薯 200 g,加水 1 L,煮沸 30 min,用两层纱布过滤,补水至1 L,加入20 g葡萄糖,自然pH,121℃下灭菌20 min。配置固体培养基时加入2 g/dL琼脂粉。
2)Aspergillus niger PCTC种子培养基:葡萄糖9 g/dL,KH2PO40.09 g/dL,MgSO4·7H2O 0.09 g/dL,(NH4)2PO40.18 g/dL,尿素 0.05 g/dL,玉米粉 0.9 g/dL,吐温-60(TW-60)1.5 g/dL;自然 pH,121 ℃下灭菌20 min。
3)LB培养基:蛋白胨 1 g/dL,酵母膏 0.5 g/dL,NaCl 1 g/dL;pH 7.0,121 ℃下灭菌 20 min, 配置固体平板时加入2 g/dL琼脂粉。抗性培养基中Amp的最终质量浓度是100 μg/mL。
4)LLB培养基:蛋白胨1 g/dL,酵母膏0.5 g/dL,NaCl 1 g/dL;pH 7.0,121 ℃下灭菌 20 min。 配置固体平板时加入2 g/dL琼脂粉。抗性培养基中zeocin的终质量浓度为25 μg/mL。
5)YPD培养基:在约800 mL的去离子水中加入酵母膏10 g,蛋白胨20 g,定容至900 mL,高压灭菌后,加入100 mL 10×D。配制固体培养基时加入2 g/dL琼脂。抗性培养基中zeocin的终质量浓度为100 μg/mL,4 ℃避光保存。
6)YPG培养基:用10×GY代替YPD培养基中的 10×D。
7)YPDS培养基:在约800 mL的去离子水中加入酵母膏10 g,蛋白胨20 g,D-山梨醇 182.2 g,定容至900 mL,高压灭菌后,加入100 mL 10×D。配制固体培养基时加入2 g/dL琼脂。抗性培养基中zeocin的终质量浓度为100 μg/mL,4℃避光保存。
8)BMMY 培养基:酵母膏 10 g,蛋白胨 20 g,加入800 mL水中,121℃下灭菌20 min,冷却至室温加入灭菌的1 mol/L磷酸钾缓冲液100 mL,10×YNB 100 mL,500×B(过滤灭菌) 2 mL,甲醇 5 mL。
1.2 实验方法
1.2.1 基因组DNA的提取和目的基因的克隆
1)黑曲霉PCTC菌体预处理:采用POD培养基平板活化黑曲霉PCTC,于28℃下培养24 h,刮取孢子配置成悬液,接种到含有50 mL种子液的250 mL摇瓶中。取培养好的种子液以相同的方法接种到含有 1.5%吐温-60(TW-60)的摇瓶中,于 200 r/min、30℃摇床中培养24 h。将数颗灭菌的玻璃珠加入到摇瓶中,置于200 r/min摇床中振荡3~4 h,取15~20 mL的菌液进行抽滤。
2)基因组DNA的提取:取上述抽滤好的菌体采用试剂盒法提取基因组DNA,参照上海生工真菌基因组DNA快速抽提试剂盒说明书提取黑曲霉PCTC的基因组DNA。
3)目的基因的获得:设计如下引物:
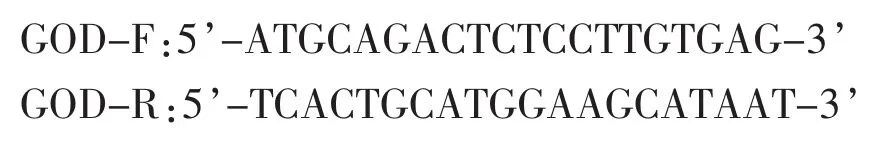
以黑曲霉基因组DNA为模板进行PCR扩增,PCR产物经纯化回收后,在T4连接酶的作用下连接到pMD18-T载体上,并转化E.coli JM109,获得重组质粒pMD18-T-GOD1,经菌落PCR鉴定获得阳性克隆子。
在克隆A.niger PCTC葡萄糖氧化酶基因时,上游引物5’端引入了位于表达载体pPICZαA的αfactor信号肽XhoI酶切位点序列 (下划线部分)和Kex2信号肽酶切位点序列(粗体部分),同时去除了葡萄糖氧化酶完整阅读框中的信号肽序列,下游引物引入了NotI酶切位点序列(下划线部分),其序列如下:
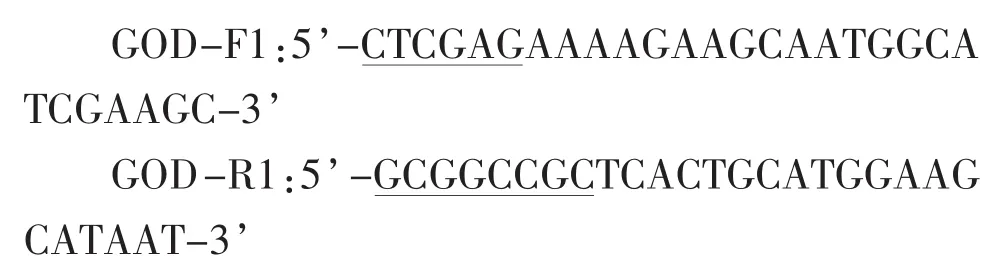
以重组质粒pMD18-T-GOD1为模板进行PCR扩增,PCR产物经纯化回收后,在T4连接酶的作用下连接到pMD18-T载体上,并转化E.coli JM109,经菌落PCR鉴定获得阳性克隆子,同时提取pMD18-T-GOD质粒送至上海生工生物工程有限公司测序进行验证。
1.2.2 重组质粒的构建 用XhoI和NotI分别将pPICZαA和pMD18-T-GOD质粒酶切。用胶回收试剂盒将pMD18-T-GOD质粒的酶切产物和pPICZαA质粒的酶切产物回收纯化。在T4连接酶的作用下构建 pPICZαA-GOD, 转化E.coli JM109,经zeocin抗性平板筛选和菌落PCR鉴定,同时送至上海生工测序验证,保存测序正确的菌株。
1.2.3 毕赤酵母的转化及筛选 采用电转化法。重组质粒pPICZαA-GOD经PmeI酶切后线性化。用线性化pPICZαA-GOD质粒电击转化毕赤酵母SMD1168感受态细胞,然后将转化细胞涂于含100 μg/mL zeocin的YPDS平板上,30℃培养3 d。随机挑选阳性菌落转接液体YPD (含100 μg/mL,以下同),培养3 d后离心收集细胞,用试剂盒提取基因组DNA并以其为模板、以5’AOX1和GOD-R1为引物进行PCR扩增以筛选、鉴定转化子SMD1168-GOD。AOX1引物如下:
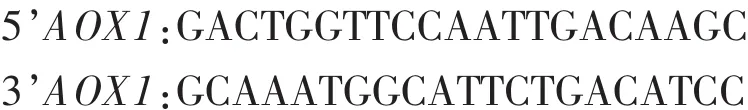
将 YPDS 平板(zeocin 100 μg/mL)上转化的单菌落用无菌牙签依次点种到含zeoein质量浓度分别为 500、1000、2 000 μg/mL 的 YPDS 平板上,30 ℃培养2~3 d,观察酵母转化子的生长情况,逐级筛选含有多拷贝目的基因的重组酵母菌株。
1.2.4 摇瓶发酵条件的优化 为了提高毕赤酵母的产酶活性,作者对pH值、诱导温度、甲醇体积分数和诱导时间等4个因素进行了优化。将筛选获得的高拷贝酵母工程菌作为生产菌株,将其接种于YPD培养基中进行活化,然后接入15 mL YPG培养基中,在30℃、200 r/min条件下培养至对数生长期,室温沉淀1~2 h,倒掉上层培养基,转入50 mL(250 mL三角瓶)液体诱导培养基BMMY,置于30℃,200 r/min摇床培养,每24小时补加体积分数1%的甲醇,诱导产酶。pH 设置为 4、5、6、7、8 五个梯度;诱导温度分别设为 15、20、25、30、35 ℃;甲醇体积分数选择0.5%、1%、1.5%、2%、3%五个浓度,72 h后测定培养物的细胞密度以及上清液的葡萄糖氧化酶。每天取样测定酶活性、OD600来确定最佳发酵时间。每组实验均设置3个平行实验来确定诱导表达时间。
1.2.5 重组葡萄糖氧化酶的分离纯化 采用饱和硫酸铵沉淀法。将重组酵母SMD1168-GOD的培养液于5 000 r/min离心5~10 min,收集上清液即为粗酶液。向粗酶液中加入饱和硫酸铵,于4℃静置过夜,10 000 r/min离心15 min,收集上清液,即为纯化后的酶液。
1.2.6 SDS-PAGE检测 利用粗酶液和纯化后的酶液进行聚丙烯酰胺凝胶电泳,经过考马斯亮蓝染色及脱色,利用凝胶成像系统进行分析。
1.2.7 酶活性的测定 酶活性测定方法采用Sigma公司和周建芹[11]的方法并加以改进:配置21 μmol/mL的双氧水标准液,反应体系中含有2.5 mL的0.21 mmol/L的邻联茴香胺溶液,0.4 mL的10 g/dL葡萄糖溶液,0.1 mL的60 U/mL的辣根过氧化物酶,再加入不同体积的21 μmol/mL双氧水,在37℃下预热5 min,加醋酸-醋酸钠缓冲溶液补到10 mL,在OD525下测定吸光度,以吸光度为横坐标,以浓度为纵坐标制作标准曲线。样品的测定用1 mL稀释好的样品代替双氧水,反应1 min测定OD值然后计算酶活性大小,酶活性单位定义为:37℃下,每分钟催化产生1 μmol的双氧水所需要的酶量做为一个酶活单位。
2 结果与分析
2.1 黑曲霉GOD基因的分析
如图1所示,用黑曲霉PCTC基因组DNA及引物GOD-F和GOD-R进行PCR扩增可获得一条约2.0 kb的DNA片段。通过测序和比对分析显示,该片段包含GOD基因,其全长为1 818 bp,编码605个氨基酸,将其序列与Genbank中其他葡萄糖氧化酶基因的序列进行对比,其相似度在92%~98%之间。
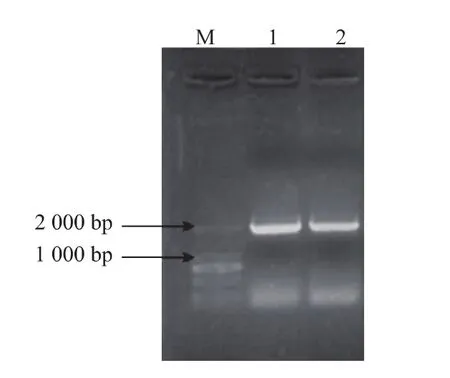
图1 黑曲霉基因组PCR扩增产物琼脂糖凝胶电泳Fig.1 Agarose gel electrophoresis of PCR amplification from PCTC
2.2 重组表达载体的构建与鉴定
将用引物GOD-F1和GOD-R1经PCR扩增并纯化回收的黑曲霉GOD基因连接到pMD18-T上。将质粒pMD18-T-GOD和pPICZαA进行双酶切以构建重组质粒,见图2。用重组质粒转化感受态大肠杆菌E.coli JM109,并在含zeocin的LB平板上筛选阳性菌落。随机挑选21个菌落,利用AOX1引物对挑取的菌落进行PCR鉴定,结果有20个菌落出现了阳性结果,显示有5.4 kb大小的条带,为目的基因和pPICZαA片段大小的总和(1.8 kb+3.6 kb)。对PCR鉴定结果正确的重组质粒进行NotI和XhoI双酶切鉴定,图3为酶切产物的电泳结果,显示有1.8 kb和3.6 kb大小的条带,分别为目的基因和表达载体片段的长度,证明GOD基因已经插入到表达载体pPICZαA中。重组质粒测序结果显示,目的基因的完整阅读框序列已正确插入到pPICZαA载体αfactor信号肽下游,重组表达载体pPICZαA-GOD构建正确。
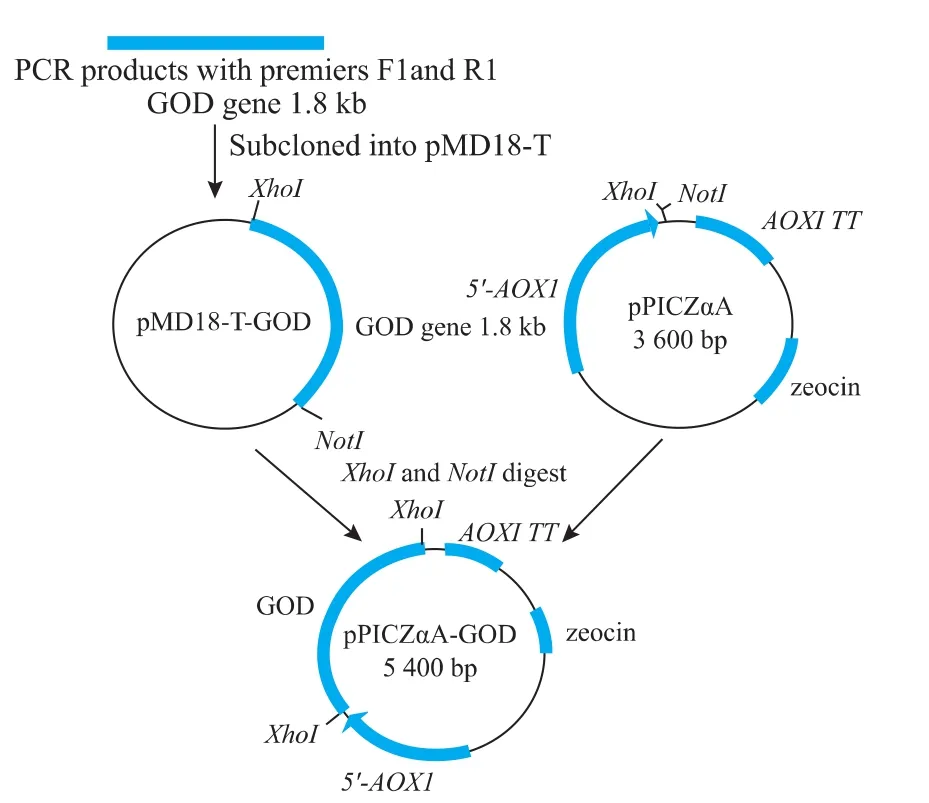
图2 重组质粒pPICZαA-GOD构建示意图Fig.2 Diagram for construction of the recombinant plasmid pPICZαA-GOD
2.3 毕赤酵母宿主菌转化与筛选
将已经线性化的重组表达载体pPICZαA-GOD电击转化,2~3 d后长出约50个酵母转化子。用牙签挑取转化单菌落依次点种在含有zeocin质量浓度分别为 500、1 000、2 000 μg/mL 的 YPDS 平板上。3 d后,酵母转化子在YPDS抗性梯度平板上的生长情况见图4。结果表明,随着zeocin质量浓度的逐级增加,长出的酵母转化子逐渐减少,a和b为长势良好的菌株,而c则为点种后未生长的菌,仍为点种前的形态。挑取在YPDS平板(zeocin 2 000 μg/mL)上生长较好的重组酵母单菌落,进行下一步的筛选鉴定。对筛选得到的若干菌株进行摇瓶复筛,通过体积分数1.0%的甲醇诱导发酵1 d获得了一株产酶最高为1.12 U/mL的菌株。
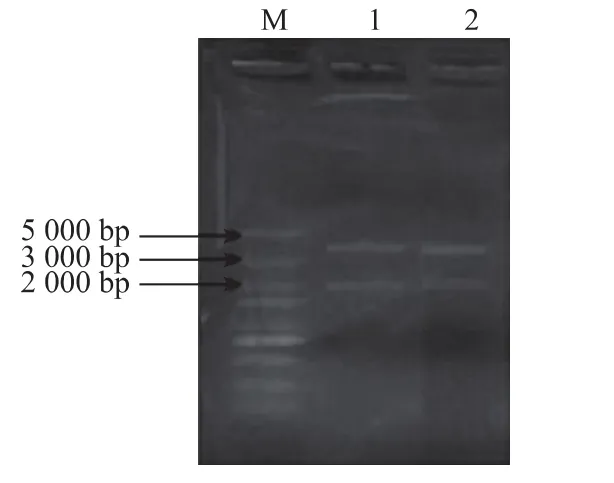
图3重组质粒pPICZαA-GOD的双酶切鉴定Fig.3 Identificationg of pPICZαA-GOD with double restriction enzymes digestion with double restriction enzymes digestion

图4 酵母转化子的抗性筛选Fig.4 Screening of P.pastoris transformants by zeocin resistance
挑取上述重组酵母的基因组,用作PCR鉴定的模板,利用上游引物5’-AOX1和下游引物GODR1进行PCR扩增,所得产物经电泳检测,结果见图5。以重组酵母基因组DNA为模板扩增的PCR产物有2.1 kb大小的条带,为葡萄糖氧化酶基因和部分AOX1启动子序列大小的总和 (1.8 kb+0.3 kb),证明目的基因已经定向整合到重组酵母菌株的基因组上。利用筛选出来的SMD1168-GOD菌株的培养物上清液做蛋白质SDS-PAGE电泳,可见一条浓染的、大小约为90 000的蛋白质条带,见图6。黑曲霉的GOD亚基相对分子质量为65 000~95 000,因此该结果提示SMD1168-GOD可以表达黑曲霉GOD蛋白。图7则为通过饱和硫酸铵沉淀法,纯化SMD1168-GOD菌株培养物上清液获得的GOD的SDS-PAGE电泳图谱。
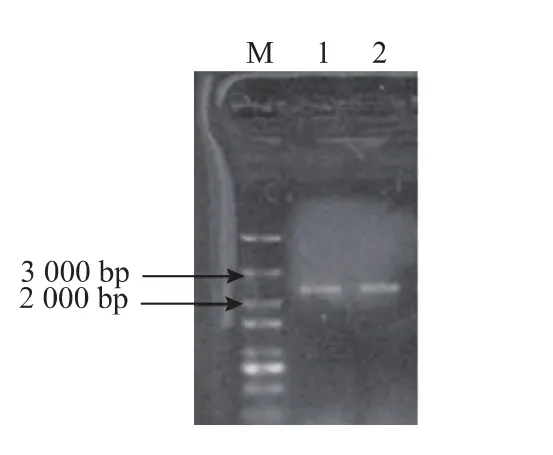
图5 重组质粒pPICZαA-GOD的PCR鉴定Fig.5 PCR identification of pPICZαA-GOD
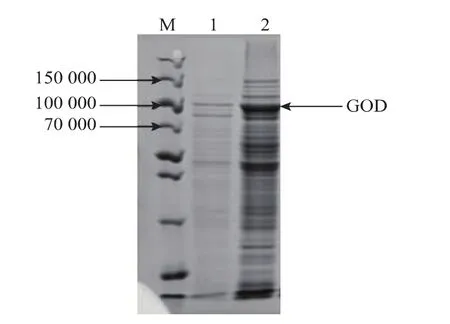
图6SMD1168-GOD菌株培养液上清液蛋白质SDS-PAGE分析Fig.6 SDS-PAGE analysis of proteins in the culture supernant of SMD1168-GOD
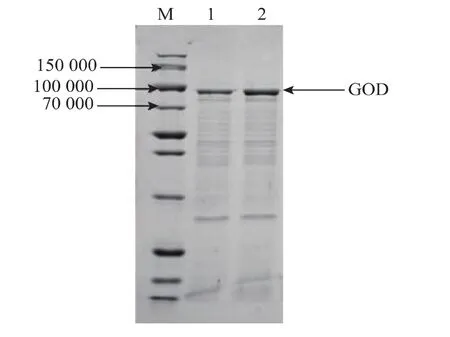
图7GOD纯化后的SDS-PAGE图谱Fig.7 SDS-PAGE analysis of the purified GOD
2.4 重组酵母发酵条件的优化
对pH值、诱导温度、甲醇浓度和诱导时间等4个因素进行了优化。结果表明,当pH为5时,重组酵母的葡萄糖氧化酶表达活力最高,见图8a。当pH继续升高时,酶活性开始下降,这可能是由于酶在弱碱性的环境下变的不稳定造成的;不同温度下诱导产酶发现,30℃下产酶较高,见图8b;甲醇体积分数对产酶也有很大影响,由图8c可以看出,随着甲醇体积分数的增加,酶活性和菌浓度显著增加,但是当其体积分数达到1.5%的时候,酶活性不再提高,且菌体的密度也开始下降,这说明甲醇体积分数过高时会抑制重组酵母产酶及生长;随着诱导时间的增加,菌体密度持续增加,酶活性也不断增加,二者存在着正相关,在第7天时葡萄糖氧化酶的活性达到最高,为17.25 U/mL,见图8d。通过发酵条件的优化,获得了最佳发酵条件为pH 5,30℃下经1.5%甲醇诱导8 d,获得的酶活最高为32 U/mL,是优化前的28倍。与Crognale等[9]利用青霉P16 GOD基因表达的酶活相比较,不仅酶活提高了96倍,且在发酵时间上减少了4 d。在表达宿主的选择上,作者采用了蛋白质缺陷型毕赤酵母SMD1168,可以将GOD分泌到细胞外,与刘虎军等[12]在毕赤酵母GS115中表达GOD相比较,不仅酶活提高了15%,且便于下游的提纯生产。与GUO等[13]比较,在培养基中补加甲醇的体积分数降低了0.5%,减少了甲醇对细胞的毒性。
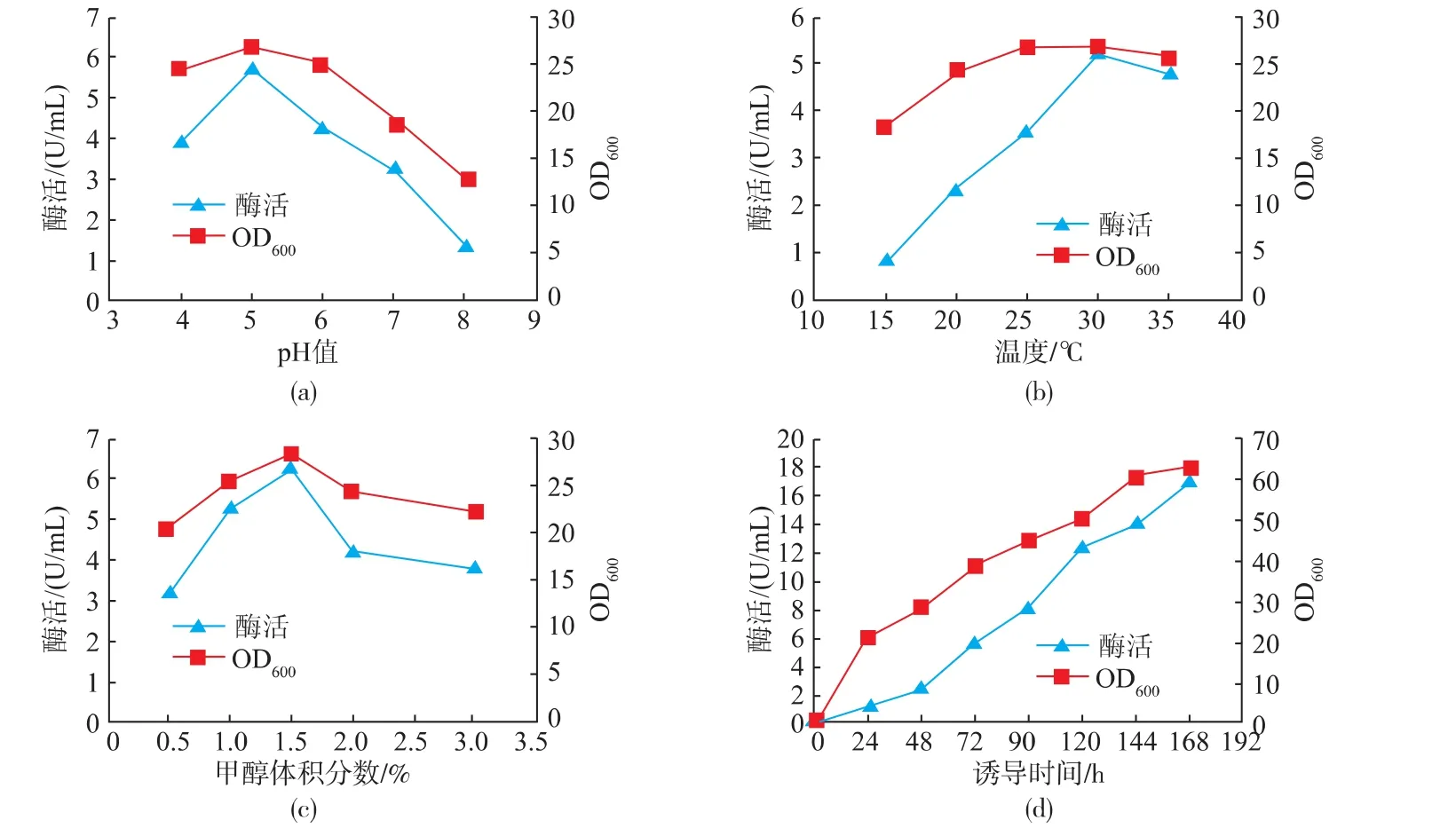
图8 重组酵母发酵条件的优化Fig.8 Optimization of fermentation conditions
3 结语
毕赤酵母的选择显著影响外源基因的表达,本研究所选择的毕赤酵母宿主SMD1168是蛋白缺陷型菌株,其Pep4基因突变,缺失蛋白水解酶活性,对表达产物降解少[14],可能会对葡萄糖氧化酶的表达量产生影响。
毕赤酵母表达系统通常以分泌表达和胞内表达两种方式来产生外源蛋白质。作者剔除了GOD基因自身长为66 bp的信号肽序列,将其成熟肽编码序列融合在α-mating factor信号肽序列下游,使得葡萄糖氧化酶能够分泌表达,便于下游的提取和纯化。另外,在α-mating factor信号肽序列上存在Ste13内肽酶酶切位点(Glu-Ala-Glu-Ala)和Kex2内肽酶酶切位点(Glu-Lys-Arg)[15],作者在设计扩增GOD基因的上游引物时,剔除了Ste13内肽酶酶切位点,而引入了Kex2内肽酶酶切位点,从而避免了表达蛋白质的N-端带有的多余氨基酸残基。
本研究获得的重组菌能够在以甲醇作为惟一碳源的培养基上生长,并在其诱导下高效表达葡萄糖氧化酶基因,在pH 5、30℃下,以1.5%的甲醇诱导7 d,其酶活性达32 U/mL。周亚凤[16]等利用pPIC9质粒将来源于黑曲霉的葡萄糖氧化酶基因在毕赤酵母 GS115中表达,酶活 30~40 U/mL;Masaki Yamaguchi[10]等利用pGAP启动子在毕赤酵母X33中表达黑曲霉9029 GOD基因,培养重组酵母14 d后,培养物上清液的平均酶活仅有1.23 U/mL。一般认为不同的表达系统和表达宿主对外源基因的表达会产生很大的影响;选择不同的启动子和信号肽也会影响外源蛋白质的表达;此外,重组酵母不同的拷贝数同样会对外源蛋白质的表达量产生巨大作用。因此,通过利用不同的表达载体和宿主菌、选用不同的启动子和信号肽、筛选高拷贝的菌株会进一步提高重组菌的GOD表达水平。
[1]BANKS J G,BOARD R G,SPARKS N H.Natural antimicrobial systems and their potential in food preservation of the future[J].Biotechnol Appl Biochem,1986,8(2-3):103-147.
[2]BONET A.Glucose oxidase effect on dough rheology and bread quality:a study from macroscopic to molecular level[J].Food Chemistry,2006,99(2):408-415.
[3]GOMEZ E,MARTINEZA,LAENCINA.Prevention ofoxidative browning during wine storage[J].Food Research International,1995,28:213-217.
[4]TZANOV T.Hydrogen peroxide generation with immobilized glucose oxidase for textile bleaching[J].Biotechnol,2002,93(1):87-94.
[5]AFSETH J,ROLLA G.Clinical experiments with a toothpaste containing amyloglucosidase and glucose oxidase[J].Caries Res,1983,17(5):472-475.
[6]CHANG G.Glucose concentration determination based on silica sol-gel encapsulated glucose oxidase optical biosensor arrays[J].Talanta,2010,83(1):61-65.
[7]KAPAT A,JUNG J K,PARK Y H.Improvement of extracellular recombinant glucose oxidase production in fed-batch culture of Saccharomyces cerevisiae:Effect of different feeding strategies[J].Biotechnology Letters,1998,20(3):319-323.
[8]CEREGHINO J L,CREGG J M.Heterologous protein expression in the methylotrophic yeast Pichia pastoris[J].FEMS Microbiol Rev,2000,24(1):45-66.
[9]CROGNALE S.Expression of Penicillium variabile P16 glucose oxidase gene in Pichia pastoris and characterization of the recombinant enzyme[J].Enzyme and Microbial Technology,2006,39:1230-1235.
[10]MASAKI Y,YUSUKE T,ATSUNORI N,et al.Secretory and continuous expression of Aspergillus niger glucose oxidase gene in Pichia pastoris[J].Protein Expression and Purification,2007,55(2):273-278.
[11]ZHOU Jianqin,CHEN Shao,WANG Jianwen.A simple and convenient method to determine the activity of glucose oxidase[J].Experimental Technology and Management,2008,25:58-60.(in Chinese)
[12]LIU Hujun,LUO Wei,FAN Xinlei,et al.Cloning and heterologous expression of glucose oxidase gene from Aspergillus niger PCTC in Pichia pastoris[J].Journal of Food Science and Biotechnology,2013,32(6):615-621.(in Chinese)
[13]Y G,F L,H Z,et al.Cloning and heterologous expression of glucose oxidase gene from Aspergillus niger Z-25 in Pichia pastoris[J].Appl Biochem Biotechnol,2010,162(2):498-509.
[14]FENG Jiankai,SUN Wanbang,CHEN Fuchao,et al.Plasmid construction and expression in Pichia pastoris SMD1168 of recombinant human interleukin-10 and purification[J].Biotechnology Bulletin,2010,212(3):168-172.(in Chinese)
[15]BRAKE A J.α-factor-directed synthesis and secretion of mature foreign proteins in Saccharomyces cerevisiae[J].Proceedings of the National Academy of Sciences,1984(15):4642-4646.
[16]ZHOU Yafeng,ZHANG Xianen,LIU Hong,et al.Cloning and expression of Aspergillus niger glucose oxidase gene in methylotrophic yeast[J].Chinese Journal of Biotechnology,2001,17(4):400-405.(in Chinese)
Expression of Aspergillus niger Glucose Oxidase Gene in Pichia pastoris SMD1168
CHEN Nan, XIAO Chengjian, FAN Xinlei, LI Hanguang, YU Xiaobin*
(School of Biotechnology,Jiangnan University,Wuxi 214122,China)
Aspergillus niger glucose oxidase (GOD) was expressed in Pichia pastoris SMD1168 with the alcohol oxidase gene promoter (AOX1).A gene of glucose oxidase from Aspergillus niger PCTC was cloned.The gene was fused to the pPICZαA plasmid which had the alcohol oxidase gene promoter (AOX1) and expressed in Pichia pastoris SMD1168.After screening by zeocin gradient resistance plate ,shake culture,and the SDS-PAGE analysis of proteins in the culture,one strain was obtained,which gave the higest enzyme activity (1.12 U/mL) after one day induction by 1%methanol.Through the optimization of the shake flask experiments,the highest enzyme activity of 32 U/mL was achieved in the flask by induction for 7 d under optimal conditions (30 ℃,200 r/min,pH 5 and 1.5%methanol).
glucose oxidase,Aspergillus niger,Pichia pastoris,heterologous expression,promoter
TQ 920.1
A
1673—1689(2017)09—0975—07
2015-03-17
中央高校基本科研业务费专项资金项目(JUSRP111A24);江苏省高校优势学科建设工程项目(111206)。
*通信作者:余晓斌(1965—),男,安徽芜湖人,工学博士,教授,博士研究生导师,主要从事发酵法生产功能食品因子方面的研究。
E-mail:xbyu@jiangnan.edu.cn
陈楠,肖成建,范新蕾,等.黑曲霉葡萄糖氧化酶基因在毕赤酵母SMD1168中的表达[J].食品与生物技术学报,2017,36(09):975-981.
猜你喜欢
饲料博览(2019年7期)2019-02-12 22:28:15
陶瓷学报(2019年5期)2019-01-12 09:17:42
猪业科学(2018年5期)2018-07-17 05:55:54
中国调味品(2017年2期)2017-03-20 16:18:25
创新作文(小学版)(2016年16期)2016-11-11 05:47:54
现代检验医学杂志(2016年5期)2016-08-20 03:17:04
中国酿造(2016年12期)2016-03-01 03:08:22
中国酿造(2016年12期)2016-03-01 03:08:20
大连工业大学学报(2015年4期)2015-12-11 04:06:50
中国科技信息(2015年2期)2015-11-16 08:18:32
