
Pd/Co3O4纳米颗粒负载于Al2O3纳米片高效催化甲烷燃烧
2017-11-01 17:29:19刘敬伟杨娜婷
物理化学学报 2017年7期
刘敬伟 杨娜婷 祝 艳
(中国科学院上海高等研究院,低碳转化科学与工程中国科学院重点实验室,上海 201210)
Pd/Co3O4纳米颗粒负载于Al2O3纳米片高效催化甲烷燃烧
刘敬伟 杨娜婷 祝 艳*
(中国科学院上海高等研究院,低碳转化科学与工程中国科学院重点实验室,上海 201210)
我们报道了一种Pd/Co3O4纳米颗粒负载于Al2O3纳米片的三元催化剂催化甲烷的高效燃烧。其中,Pd/Co3O4负载于碱性氧化铝的复合材料活化甲烷C―H键的能力比SiO2、ZrO2和CeO2以及酸性和中性Al2O3为载体时更强,这是因为Pd/Co3O4/碱性Al2O3拥有更多的氧空穴和吸附氧,对催化剂催化甲烷燃烧有较好的影响。尽管在5%(体积分数)的水蒸气存在下,其催化性能有一定的失活,但在移除水蒸气时,其催化性能可以快速恢复至最佳状态。在模拟真实汽车尾气的氛围下,Pd/Co3O4/碱性Al2O3依然具有较好的催化甲烷燃烧性能,在400 °C时可以催化甲烷完全转化。
Pd/Co3O4;碱性;甲烷氧化;活性;稳定性
1 Introduction
Natural gas, of which methane is the major component, is widely used in power generation, vehicle fuel and other heating applications1. Methane is a very stable and symmetrical molecule containing four C―H bonds2. The breaking of the C―H bond is thermodynamically unfavorable and the energyof methane activation is higher than those of higher hydrocarbons3. Therefore, the conversion of methane, such as oxidative coupling of methane for ethane and ethylene4,methane dry reforming for CO and H25, and partial oxidation of methane for methanol6, need carrying out at high temperature even if in the presence of catalysts. The catalytic combustion of methane, as one of methane conversion, cannot be happened at low temperature and the complete methane combustion is obtained below at least 400 °C in the very active catalysts7−9.However, the complete oxidation of unburned methane from exhaust streams is significant for that the greenhouse effect of methane is more than 20 times than that of carbon dioxide, and the incomplete oxidation could bring about various nitrogen oxides, carbon monoxide, and soot emissions10. With given more attention to environment and health, the high catalytically active and stable catalysts thus are highly desirable to, in principle, emission limit of methane to the atmosphere, and simultaneously reduce NOx, CO and soot emissions to ultra-low levels11. Current popular catalysts for methane catalytic combustion are noble metals and metal oxides12,13.Several crucial factors need to be considered in developing new catalysts for catalytic combustion of methane, such as high sintering rates, high cost of noble metals, water and sulfur poisoning14,15. Palladium-based catalysts are known as the very effective catalysts for catalytic methane oxidation, and unfortunately, they tend to deactivate because of sintering at high temperature14,16. Other catalysts based on oxides such as transition metal oxides, perovskite oxides and hexaaluminate oxides show low catalytic activity, with the complete conversion of methane observed only above 400 °C17−19.
In terms of Pd-based catalysts, many efforts have been made to investigate the influence of the metal-oxide support on the activity and the stability of the palladium catalyst, the impact of the support is mainly credited to palladium dispersion, oxygen mobility, and the Pd-support interaction20,21. The metal oxides not only work as supports but also function as electronic modulators to contribution spillover and adsorption sites22,23.Co3O4with typical spinel-structure transition metal oxide is an important support for Pd-based catalysts in the activation of methane, since there is a strong interaction between the Co3O4unit cell and the PdO unit cell24,25. Aluminum oxides(especially γ-Al2O3), with high surface area, moderate chemical activity, and low cost, are also widely employed as supports for methane combustion26,27.
In view of the virtue of Co3O4and Al2O3as supports, herein,we produce an efficient catalyst such Pd/Co3O4/Al2O3through inlaying Pd/Co3O4nanoparticles in Al2O3nanosheets for catalytic oxidation of methane. Pd/Co3O4inlaid in Al2O3catalysts exhibit higher catalytic ability compared to SiO2, ZrO2and CeO2as supports. Pd/Co3O4supported on alkaline Al2O3nanosheets is more efficient than Pd/Co3O4supported on acidic or neutral Al2O3for methane oxidation. Finally,Pd/Co3O4/alkaline Al2O3catalyst exhibited superior performance for methane oxidation in the presence of water vapor and a mixed gas similar to the exhaust of lean-burn natural gas engine, which indicates that this catalyst would be very interesting for this application.
2 Experimental
2.1 Chemicals and reagents
Cobalt (II) acetate tetrahydrate (Co(Ac)2·4H2O, 99.5%),ethylene glycol (EG, 99%), sodium carbonate anhydrous(Na2CO3, 99.8%), and potassium tetrachloropalladate (K2PdCl4,98%), were purchased from Aladdin reagent Co., Ltd. Acidic Al2O3(FCP, 200−300 mesh), neutral Al2O3(FCP, 200−300 mesh), alkaline Al2O3(FCP, 200-300 mesh) (the acid-base property of these Al2O3explained by the CO2temperature-program desorption was shown in Fig.S1(Supporting Information)), SiO2(AR), ZrO2(AR), CeO2(AR),were purchased from Sinopharm Chemical Reagent Co., Ltd.All of the chemicals were used directly without further treatment except metal oxides. Metal oxides were pretreated at 900 °C for 12 h.
2.2 Catalysts preparation
Pd/Co3O4nanoparticals: Co(Ac)2·4H2O (5 mmol) was dissolved in ethylene glycol (15 mL) at 80 °C under vigorous stirring and argon flow. Na2CO3aqueous solution (0.2 mol·L−1,50 mL) was then added to the mixture drop-wisely and purple precipitate was gained, which was further aged for 10 min at 80 °C. After that, to maintain the metal loading on the Co3O4supported catalysts, 10 mL aqueous solution of appropriate amount of Na2PdCl4was then added to the aforementioned solution. The resulting solution was further aged at that temperature for 1.5 h. After the solution was cooled to room temperature, a dark purple solid precipitate was separated by centrifugation with successive repeated washing with water and ethanol. After the precipitate was dried at 60 °C in vacuum, the obtained Pd/cobalt hydroxycarbonate was calcined to produce Pd/Co3O4nanoparticles in air at 300 °C for 3 h.
Pd/Co3O4and Al2O3(or SiO2, ZrO2, CeO2) were co-added into 15 mL of ethanol and the mixture stirred overnight.Solvent was then centrifugation and the powder dried at 120 °C overnight, ground to a particle size below 150 μm and calcined in air at 500 °C for 5 h.
2.3 Catalysts characterization
Powder X-ray diffraction (XRD) of the samples was recorded on an Ultima IV, made in Japan, performed with Cu Kαradiation (0.15418 nm). The TEM image, HAADF and element mapping images were acquired from using the transmission electron microscope (G2 F20 S-Twin, FEI,America) equipped with energy dispersive X-ray spectroscopy(EDS) at an accelerating voltage of 200 kV. The binding energies of surface species on the catalysts were determined by X-ray photoelectron spectroscopy (XPS, Thermo Scientific K-Alpha, America) using Al Kα(hv = 1486.6 eV) as the excitation source and the pass energy is 145 eV. The N2sorption experiments were done with physical adsorption analyzer (TriStar II 3020, Micromeritics, Germany). All the samples were degassed at 200 °C for 6 h under vacuum prior to measurement. The surface areas were calculated using the Brunauer-Emmett-Teller (BET) method. Methane temperature-programmed reduction (CH4-TPR) and oxygen temperature-programmed desorption (O2-TPD) experiments were carried out on a chemical adsorption analyzer (Autochem II 2920, Micromeritics, America) with a mass spectrograph(Cirrus 2). Before TPR measurement, 50 mg of catalyst was pretreated in an air flow at 300 °C for 2 h. After being cooled at the same atmosphere to 60 °C, the sample was exposed to a flow (50 mL·min−1) of 10% (volume fraction) CH490%(volume fraction) He mixture and kept at 60 °C for 1 h, finally the pretreated sample was heated from 60 to 800 °C at a ramp of 10 °C·min−1; and before TPD measurement, 200 mg of catalyst was pretreated in an He flow at 300 °C for 2 h. After being cooled at the same atmosphere to 60 °C, the sample was exposed to a flow (50 mL·min−1) of 1% (volume fraction) O299% (volume fraction) He mixture and kept at 60 °C for 2 h,and then turned to a He flow for 2 h, finally the pretreated sample was heated from 60 to 800 °C at a ramp of 10 °C·min−1.
2.4 Catalytic evaluation
Catalytic combustion of methane was measured using a conventional quartz tubular reactor (diameter, 6 mm; length,500 mm) at atmospheric pressure. The reagent gas mixture (1%(volume fraction) CH4, 10% (volume fraction) O2and 89%(volume fraction) N2) was led over catalysts (40−60 mesh, 0.1 g) at a flow rate of 100 mL·min−1, equivalent to a space velocity (SV) of 30000 mL·g−1·h−1. The reaction of methane combustion was heated from 20 to 450 °C at a ramp of 5 °C·min−1. The reaction products were analyzed by micro gas chromatograph (INFICON 3000) equipped with MS5A and Plot Q columns (for separation of H2, CO, N2, CO2, O2, CH4,C2H4, and C2H6) and TCD detector.
3 Results and discussion
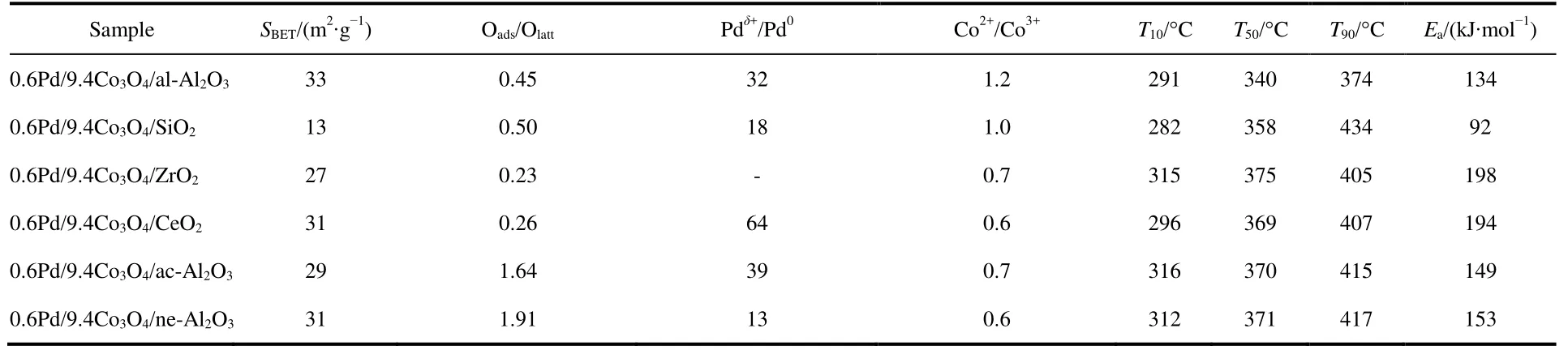
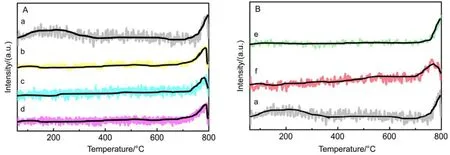
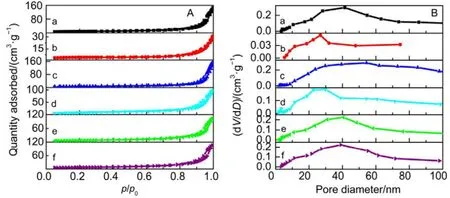
Catalytic performance toward methane oxidation was evaluated by obtaining methane conversion vs temperature profiles of Pd/Co3O4nanoparticles loaded on different oxide supports (0.6% (mass fraction) Pd and 9.4% (mass fraction)Co3O4supported on alkaline Al2O3(denoted as al-Al2O3), SiO2,ZrO2or CeO2, respectively) for comparison. All the catalysts showed 100% selectivity toward CO2, and no CO was observed during the reaction. Pd/Co3O4/al-Al2O3catalyst is more effective than other three catalysts for methane oxidation. As shown in Fig.1A, methane can be lighted off at 250 °C and completely oxidized at 400 °C over Pd/Co3O4/al-Al2O3catalyst at a flow rate of 100 mL·min−1with 1% (volume fraction) CH4and 10% (volume fraction) O2balanced with N2. Although methane can be ignited over Pd/Co3O4/SiO2catalyst at 250 °C,the complete oxidation of methane cannot be obtained at 400 °C, that is, only 70% methane was oxidized at 400 °C.Methane needs higher energy to be activated and converted over Pd/Co3O4/ZrO2and Pd/Co3O4/CeO2catalysts, where methane was initially activated at above 275 °C and less than 90% methane was converted at 400 °C. The kinetics of methane oxidation at the initial stages of reaction was studied,and a good linear relation with data reveals the pseudo first-order model of methane. It is noted that the reaction rate of Pd/Co3O4/SiO2catalyst is faster than other ones at a low operating temperature (< 325 °C), while the conversion of methane over Pd/Co3O4/al-Al2O3catalyst is the highest among the four catalysts at above 325 °C (Fig.1B).
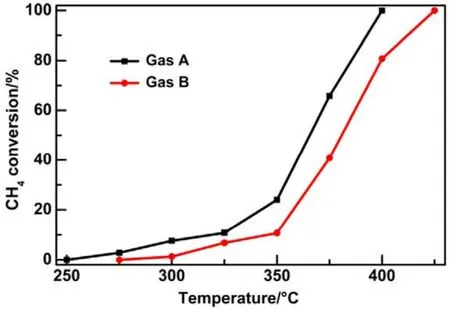
Afterwards, we studied the influence of acidic, neutral and alkaline properties of Al2O3substrate on the catalytic behavior of Pd/Co3O4for catalytic oxidation of methane. From Fig.1C,Pd/Co3O4/acidic Al2O3and Pd/Co3O4/neutral Al2O3catalysts did not give the complete oxidation of methane at 425 °C. T90of the two catalysts are 40 °C higher than the case of Pd/Co3O4/alkaline Al2O3. The measured T10and T90temperatures follow the order of Pd/Co3O4/alkaline Al2O3 The high efficient activity of Pd/Co3O4/al-Al2O3catalyst is partially associated with its strong redox property, which has been confirmed by the CH4temperature-program reduction studies. The CH4-TPR results are shown in Fig.2, and the consumption peaks of CH4and the formation of CO2, H2O, CO,and H2are detected. For Pd/Co3O4/al-Al2O3in Fig.2A, the first consumption peak of CH4at 232 °C is attributed to the reduction process of the PdOxwith CH4toward Pd0. The second one at 420 °C is related with the reduction reaction of Co3O4with CH4from Co3+to Co2+. The third consumption peak at 525 °C corresponds to the reduction process of Co3O4with CH4from Co2+to Co0and the reforming reaction of CH4and CO2or H2O under the catalysis of Co0or Pd0species, due to the formation of CO2, H2O, CO and H228. The fourth weak peak centered 537 °C can be attributed to the cracking of CH4.For the Pd/Co3O4/SiO2in Fig.2B, the reduction peak of Pdδ+to Pd0shifts to 281 °C and the reduction peak of Co3+to Co2+moves to 313 °C, respectively. The peak at 706 °C with the formation of H2O and trace CO2and CO might result from the reduction of Co2+to Co0. In terms of Pd/Co3O4/ZrO2in Fig.2C,the peaks of Pdδ+to Pd0and Co3+to Co2+move to 304 and 381 °C, and the peak at 393 °C is attributed to the reduction of Co2+to Co0and the reforming of methane. The peak at 669 °C is assigned to the cracking of methane. As to Pd/Co3O4/CeO2shown in Fig.2D, the four peaks are observed at 290, 415, 578 and 676 °C, respectively, due to the reduction of Pdδ+to Pd0,Co3+and Co2+to Co0, cracking of methane, and the reforming of methane. According to reduction temperatures, the redox ability of the catalysts are followed a descended sequence:Pd/Co3O4/al-Al2O3< Pd/Co3O4/SiO2< Pd/Co3O4/CeO2 Fig.1 (A) The catalytic performance and (B) the corresponding kinetic rate data of Pd/Co3O4 nanoparticles supported on (a) alkaline Al2O3,(b) SiO2, (c) ZrO2 and (d) CeO2 for catalytic oxidation of methane, respectively; (C) the light-off profiles of catalytic oxidation of methane over (e) Pd/Co3O4/acidic Al2O3 (denoted as ac-Al2O3), (f) Pd/Co3O4/neutral Al2O3 (denoted as ne-Al2O3), and(a) Pd/Co3O4/alkaline Al2O3, respectively, (D) the corresponding kinetic rate data for methane oxidation. As we known, the adsorbed oxygen species are more active than the lattice oxygen for catalytic oxidation of methane. The more effective activity of alkaline Al2O3loaded by Pd/Co3O4for methane oxidation is also attributed to the more adsorbed oxygen species of this catalyst. From the analysis of O2-TPD data shown in Fig.3, the peaks below 400 °C are attributed to the desorption of surface adsorbed oxygen species (such as O2−and O−), while the peaks beyond 700 °C are assigned to the desorption of lattice oxygen species. The abroad band of Pd/Co3O4/alkaline Al2O3at about 200−400 °C points out thepresence of reactive oxygen species on the surface of this catalyst, while no obvious signal of adsorbed oxygen species were found on other five catalysts, indicating that alkaline Al2O3benefits for the formation of adsorbed oxygen species,which ultimately promotes the activation of methane on Pd/Co3O4/alkaline Al2O3catalyst. Table 1 BET specific surface area (SBET) from N2 absorption isotherms, the molar ratios of adsorbed oxygen/lattice oxygen (Oads/Olatt),Pdδ+/Pd0, and Co2+/Co3+ detected from XPS, the temperatures for 10%, 50% and 90% conversion of methane (T10, T50 and T90),and apparent activation energy (Ea). Fig.2 CH4 temperature-program reduction spectra of (A) Pd/Co3O4/ alkaline Al2O3, (B) Pd/Co3O4/SiO2,(C) Pd/Co3O4/ZrO2, (D) Pd/Co3O4/CeO2, (E) Pd/Co3O4/acidic Al2O3, and (F) Pd/Co3O4/neutral Al2O3. Fig.3 O2 temperature-program desorption spectra of (a) Pd/Co3O4/alkaline Al2O3, (b) Pd/Co3O4/SiO2, (c) Pd/Co3O4/ZrO2,(d) Pd/Co3O4/CeO2, (e) Pd/Co3O4/acidic Al2O3, and (f) Pd/Co3O4/neutral Al2O3. Besides the comparison of the redox property and active oxygen species of the catalysts, we now turn to consider the surface compositions, which were showed in Fig.S2 and Table S1. Pd 3d binding energy29,30of the five samples except Pd/Co3O4/ZrO2(note: Pd peaks are overlapped by Zr peaks)show no shift toward high or low binding energy and the cationic Pdδ+dominates the surface of samples, as shown in Fig.4A, while the presence of trace Pd0is in Pd 3d spectra of the five samples. The molar ratios of Pdδ+/Pd0of these samples were calculated and listed in Table 1. Here the ratio value of Pdδ+/Pd0seems to be not directly related with the catalytic activity, though PdO is more active than Pd for methane oxidation. Fig.4B presents the Co 2p spectra and it is found that the occurrence of satellite structure leads to the deconvolution of Co 2p1/2and 2p3/2excitations. The peaks located at 781.8 eV are attributable to Co2+, and the peaks at 779.9 eV are assigned to Co3+29,31,32, which revealed the presence of Co2+and Co3+of all samples, demonstrating that CoO and Co2O3dominated the surface of the samples. The molar ratios of Co2+/Co3+listed in Table 1 indicated that Pd/Co3O4/al-Al2O3gave the highest value of Co2+/Co3+among these samples. Co2+is considered to be the source of oxygen vacancy and raising the relative amount of Co2+on the catalysts’ surface benefits for increasing the amount of oxygen vacancy and then benefits for methane conversion7,13. Thus the high concertration of Co2+might be one reason to explain the high activity of Pd/Co3O4/al-Al2O3.However, the detailed order for Co2+/Co3+value of these catalysts follows Pd/Co3O4/al-Al2O3> Pd/Co3O4/SiO2>Pd/Co3O4/ZrO2> Pd/Co3O4/CeO2, and Pd/Co3O4/al-Al2O3>Pd/Co3O4/acidic Al2O3> Pd/Co3O4/neutral Al2O3, which is almost but no complete agreement with the experimental results obtained in methane oxidation. Actually, the activity is attributed to the collective contribution from all factors rather than only one. Fig.4 (A) Pd 3d and (B) Co 2p of (a) Pd/Co3O4/alkaline Al2O3, (b) Pd/Co3O4/SiO2, (c) Pd/Co3O4/ZrO2, (d) Pd/Co3O4/CeO2,(e) Pd/Co3O4/acidic Al2O3, and (f) Pd/Co3O4/neutral Al2O3. Fig.5 (A) Nitrogen adsorption-desorption isotherms of (a) Pd/Co3O4/alkaline Al2O3, (b) Pd/Co3O4/SiO2, (c) Pd/Co3O4/ZrO2,(d) Pd/Co3O4/CeO2, (e) Pd/Co3O4/acidic Al2O3, and (f) Pd/Co3O4/neutral Al2O3; (B) the corresponding pore size distribution. The BET surface areas and pore characters of these catalysts were determined from N2sorption measurements, which are shown in Fig.5 and Table 1. The BET surface area of these six catalysts (except SiO2) seems to be very close, suggesting that their catalytic activity seems to be not directly related with their specific surface area. We also investigated the effect of loading amount of Pd and Co3O4on alkaline Al2O3on the catalytic performance of methane oxidation. When the loading amount of Pd/Co3O4is fixed to 10% (mass fraction), it is found that as the amount of Pd loaded on Co3O4is turned into 3% (mass fraction), 5%(mass fraction), 6% (mass fraction) and 10% (mass fraction),respectively, a interesting phenomenon is observed that 0.6Pd/9.4Co3O4/Al2O3(the loading amount of Pd is 6% (mass fraction)) catalyst showed superior catalytic performance(Fig.6A) to 0.3Pd/9.7Co3O4/Al2O3, 0.5Pd/9.5Co3O4/Al2O3and 1.0Pd/9.0Co3O4/Al2O3. As shown in Fig.6B, when the amount of Pd loaded on Co3O4is fixed to 5% (mass fraction),0.75Pd/14.25Co3O4/Al2O3(15% Pd/Co3O4on Al2O3) and 1.0Pd/19.0Co3O4/Al2O3(20% Pd/Co3O4on Al2O3) are similar light-off curves of catalytic oxidation of methane, yet which are higher activity than 0.5Pd/9.5Co3O4/Al2O3(10% Pd/Co3O4on Al2O3) catalyst. When the amount of Pd loaded on Co3O4is fixed to 6% (mass fraction), for the three catalysts, such as 0.9Pd/14.1Co3O4/Al2O3(15% (mass fraction) Pd/Co3O4on Al2O3) and 1.2Pd/18.8Co3O4/Al2O3(20% (mass fraction)Pd/Co3O4on Al2O3), the temperatures of methane conversion are close over the two catalysts under same evaluation condition, which is lower than that of 0.6Pd/9.4Co3O4/Al2O310% (mass fraction) Pd/Co3O4on Al2O3) (Fig.6B). Thus, from the above results, even if the weight percent of Pd is above 0.6%, 0.6Pd/9.4Co3O4/al-Al2O3always gave the highest catalytic activity among these catalysts. Fig.6 (A, B) Catalytic results of methane oxidation over Pd/Co3O4/al-Al2O3 with different loading amount of Pd and Co3O4; (C) cycle of heating and cooling cycle profiles of methane conversion against the temperature for 0.6Pd/9.4Co3O4/al-Al2O3; (D) catalytic stability of 0.6Pd/9.4Co3O4/al-Al2O3 at 380 °C for 50 h (the upper panel), and 500 °C for 50 h (the lower panel); (E) 5% (volume fraction) vapor water effect on the catalytic performance of 0.6Pd/9.4Co3O4/al-Al2O3 at 380 °C. Hence 0.6Pd/9.4Co3O4/al-Al2O3catalyst holds promising in its utility as a type of Pd-based catalysts that offer high activity for methane oxidation. Especially, this catalyst displayed a long-term stability without an obvious decline in activity whatever cycles and time-on-stream experiments. When the temperature was ramped from 250 to 400 °C and then cooled back to 250 °C, the light off curve for upward ramp was virtually identical to one for downward ramp (Fig.6C). The second heating and cooling cycle test showed no significant deactivation compared to the first cycle (Fig.6C). Moreover,0.6Pd/9.4Co3O4/al-Al2O3catalyst exhibited a high sintering resistance at high temperature. When the temperature was kept at 380 °C, noted that the conversion of methane is not complete, the conversion of methane can always be maintained about 97% for at least 50 hours (the upper panel of Fig.6D).Even if the temperature was fixed at high temperature such 500 °C, the 100% conversion of methane over 0.6Pd/9.4Co3O4/al-Al2O3can be obtained when the reaction time was up to 50 hours (the lower panel of Fig.6D).Especially, the effect of moisture on the catalytic performance of this catalyst for methane oxidation was examined. To investigate the catalytic performance of the catalysts for wet methane combustion, 5% (volume fraction) of water vapor was introduced by a calibrated water pump and vaporized in the heated gas feed line before entering the reactor. It is found that Pd/Co3O4/alkaline Al2O3catalyst can almost restore to its initial value in the absence of water when 5% (volume fraction) water was cut off and the reaction temperature was at 380 °C,although a decrease from 97% to 80% in activity occurred when water was introduced to the reaction system (Fig.6E). Interestingly, the structure of Pd/Co3O4/al-Al2O3catalyst has very little change and no significant aggregation after catalytic reaction when compared to the fresh one (Fig.7). For fresh catalyst with the average PdO particle size of 6 nm, we can see the (220) and (222) lattice fringe with interplanar spacing of 0.199 and 0.163 nm (calculated from the fast Fourier transformed pattern of the red circled area shown in Fig.7B and 7E) on the PdO (Fig.7B). The lattice spacing of 0.232 nm can be assigned to the (111) lattice fringe of Co3O4. The energy-dispersive X-ray spectroscopy (EDS) mapping analysis(Fig.7C) clearly points to the distribution ranges of Pd (red) and Co (blue) in the nanoparticle, revealing the homogenous distribution of Pd and Co in Al2O3support. After catalytic test,TEM studies offer further insight into the structural information of spent catalyst. For spent catalyst with the average PdO particle size of 10 nm in Fig.7(D−F), Pd and Co are coexisted on the support and the contact of PdO and Co3O4is still intimate, which indeed ensures a high resistance to sintering of active sites at thermal condition. Fig.7 (A) TEM and (B) HRTEM images (inset: the fast Fourier transformed pattern of the red circled area, spots 1, 2 and 3 represent the Co3O4(111), PdO (220) and PdO (222), respectively) of fresh 0.6Pd/9.4Co3O4/al-Al2O3 catalyst and (C) corresponding EDS elemental mapping.(D) TEM and (E) HRTEM images (inset: the fast Fourier transformed pattern of the red circled area, spots 1, 2 and 3 represent the Co3O4 (111),PdO (220) and PdO (220), respectively) of spent 0.6Pd/9.4Co3O4/al-Al2O3 catalyst and (F) corresponding EDS elemental mapping. Fig.8 Catalytic performance of 0.6Pd/9.4Co3O4/al-Al2O3 under conditions simulating the mixture gases of exhaust of engines of natural gas vehicles. Further, to evaluate the catalytic performance of 0.6Pd/9.4Co3O4/al-Al2O3catalyst in methane oxidation of gas exhaust of an engine of natural gas, two different mixed gases similar to the exhaust of lean-burn natural gas engine were used32: one is composed with CH4: 0.20%, O2: 10%, CO2: 15%,H2O: 5% and N2as the balance at space velocity of 30000 mL·g−1·h−1, and the other is mixed with CH4: 0.20%, O2: 10%,NO: 0.15%, H2O: 5% balanced with N2. In the case of CH4:0.20%, O2: 10%, CO2: 15%, H2O: 5%, and N2: 69.80%,0.6Pd/9.4Co3O4/al-Al2O3catalyst gave rise to a complete conversion of methane at 400 °C. From Fig.8, in the mixture of CH4: 0.20%, O2: 10%, NO: 0.15%, H2O: 5%, and N2: 84.65%,100% conversion of methane can be achieved over 0.6Pd/9.4Co3O4/al-Al2O3catalyst at 425 °C. The results reflect that Pd/Co3O4/Al2O3catalyst shows higher activity than Pd/Al2O3reported above 500 °C under the conditions simulating the typical gas composition of the exhaust of engines of natural gas33. In summary, we report an efficient catalyst composed of ternary components via Pd/Co3O4nanoparticles inlaid in alkaline Al2O3nanosheets for catalytic oxidation of methane.The high adsorbed oxygen species concentration and strong redox properties appear to enhance the oxidation activity of Pd/Co3O4/al-Al2O3. The intimate contact of Pd/Co3O4with Al2O3ensures a high resistance toward sintering at thermal condition. The addition of 5% (volume fraction) water did not seriously affect the catalytic activity of Pd/Co3O4/al-Al2O3and the activity can restore to its initial value in the absence of water when the water was cut off. The excellent catalytic behavior for methane oxidation of Pd/Co3O4/al-Al2O3highlights the importance of pursuing the ternary recipes for real-world application. Acknowledgment: We thank Dr. Andrew Haslett from ETI and Prof. Graham J. Hutchings from Cardiff University for helpful discussions. Supporting Information: available free of charge via the internet at http://www.whxb.pku.edu.cn. (1) Choudhary, T. V.; Banerjee, S.; Choudhary, V. R. Appl. Catal. A 2002,234, 1. doi: 10.1016/S0926-860X(02)00231-4 (2) Gélin, P.; Primet, M. Appl. Catal.: B 2002, 39, 1.doi: 10.1016/S0926-3373(02)00076-0 (3) Tang, P.; Zhu, Q.; Wu, Z.; Ma, D. Energy Environ Sci. 2014, 7, 2580.doi: 10.1039/C4EE00604F (4) Song, J.; Sun, Y.; Ba, R.; Huang, S.; Zhao, Y.; Zhang, J.; Sun, Y.;Zhu, Y. Nanoscale 2015, 7, 2260. doi: 10.1039/c4nr06660j (5) Yuan, K.; Zhong, J.; Zhou, X.; Xu, L.; Bergman, S. L.; Wu, K.; Xu,G.; Bernasek, S. L.; Li, H.; Chen, W. ACS Catal. 2016, 6, 4330. doi:10.1021/acscatal.6b00357 (6) Chen, L.; Zhang, X.; Huang, L.; Lei, L. Chem. Eng. Process 2009,48, 1333. doi:10.1016/j.cep.2009.06.007 (7) Wang, Q.; Peng, Y.; Fu, J.; Kyzas, G. Z.; Billah, S. M. R.; An, S.Appl. Catal. B 2015, 168−169, 42. doi: 10.1016/j.apcatb.2014.12.016 (8) Najjar, H.; Batis, H.; Lamonier, J. F.; Mentré, O.; Giraudon, J. M.Appl. Catal. A 2014, 469, 98. doi: 10.1016/j.apcata.2013.09.014 (9) Okal, J.; Zawadzki, M. Appl. Catal. A 2013, 453, 349.doi: 10.1016/j.apcata.2012.12.040 (10) Massias, A.; Diamantis, D.; Mastorakos, E.; Goussis, D. Combust.Theor. Model. 1999, 3, 233. doi: 10.1088/1364-7830/3/2/002 (11) Ciuparu, D.; Lyubovsky, M. R.; Altman, E.; Pfefferle, L. D.; Datye,A. Catal. Rev. 2002, 44, 593. doi: 10.1081/CR-120015482 (12) Zou, X.; Rui, Z.; Song, S.; Ji, H. J. Catal. 2016, 338, 192. doi:10.1016/j.jcat.2015.12.031 (13) Sun, Y.; Liu, J.; Song, J.; Huang, S.; Yang, N.; Zhang, J.; Sun, Y.;Zhu, Y. ChemCatChem 2016, 8, 540. doi: 10.1002/cctc.201000407 (14) Niu, F.; Li, S.; Zong, Y.; Yao, Q. J. Phys. Chem. C 2014, 118, 19165.doi: 10.1021/jp504859d (15) Di, G.; Melaet, G.; Kruse, N.; Liotta, L. F.; Pantaleo, G.; Venezia, A.M. Chem. Commun. 2010, 46, 6317. doi: 10.1039/C0CC00723D (16) Hoffmann, M.; Kreft, S.; Georgi, G.; Fulda, G.; Pohl, M. M.;Seeburg, D.; Berger-Karin, C.; Kondratenko, E. V.; Wohlrab, S. Appl.Catal. B 2015, 179, 313. doi: 10.1016/j.apcatb.2015.05.028 (17) Li, H.; Lu, G.; Qiao, D.; Wang, Y.; Guo, Y. Catal. Lett. 2011, 141,452. doi: 10.1007/s10562-010-0513-y (18) Cimino, S.; Lisi, L.; Pirone, R.; Russo, G.; Turco, M. Catal. Today 2000, 59, 19. doi: 10.1016/S0920-5861(00)00269-8 (19) Li, S.; Wang, X. Catal. Commun. 2007, 8, 410.doi:10.1016/j.catcom.2006.07.011 (20) Kucharczyk, B.; Tylus, W. Catal. Today 2008, 137, 324.doi: 10.1016/j.cattod.2008.05.018 (21) Wang, X.; Guo, Y.; Lu, G.; Hu, Y.; Jiang, L.; Guo, Y.; Zhang, Z.Catal. Today 2007, 126, 369. doi: 10.1016/j.cattod.2007.06.011 (22) Yoshida, H.; Nakajima, T.; Yazawa, Y.; Hattori, T. Appl. Catal. B 2007, 71, 70. doi:10.1016/j.apcatb.2006.08.010 (23) Ercolino, G.; Grodzka, A.; Grzybek, G.; Stelmachowski, P.;Specchia, S.; Kotarba, A. Top. Catal. 2017, 3, 333.doi: 10.1007/s11244-016-0620-0 (24) Ercolino, G.; Grzybek, G.; Stelmachowski, P.; Specchia, S.; Kotarba,A.; Specchia, V. Catal. Today 2015, 257, 66.doi: 10.1016/j.cattod.2015.03.006 (25) Hu, L.; Peng, Q.; Li, Y. ChemCatChem 2011, 3, 868.doi: 10.1002/cctc.201000407 (26) Xu, J.; Ouyang, L.; Mao, W.; Yang, X.; Xu, X.; Su, J.; Zhuang, T.;Li, H.; Han, Y. ACS Catal. 2012, 2, 261.doi: 10.1021/acscatal.6b00357 (27) Cargnello, M.; Delgado, J. J.; Hernandez, J. C.; Bakhmutsky, K.;Montini, T.; Calvino, J. J.; Gorte, R. J.; Fornasiero, P. Science 2012,337, 713. doi: 10.1126/science.1222887 (28) Wu, Z.; Deng, J.; Liu, Y.; Xie, S.; Jiang, Y.; Zhao, X.; Yang, J.;Arandiyan, H.; Guo, G.; Dai, H. J. Catal. 2015, 332, 13.doi: 10.1016/j.jcat.2015.09.008 (29) Giraudon, J. M.; Elhachimi, A.; Leclercq, G. Appl. Catal. B 2008, 84,251. doi: 10.1016/j.apcatb.2008.04.023 (30) Liu, Y.; Dai, H.; Deng, J.; Xie, S.; Yang, H.; Tan, W.; Han, W.;Jiang, Y.; Guo, G. J. Catal. 2014, 309, 408.doi: 10.1016/j.jcat.2013.10.019 (31) Xie, S.; Deng, J.; Zang, S.; Yang, H.; Guo, G.; Arandiyan, H.; Dai, H.J. Catal. 2014, 322, 38. doi: 10.1016/j.jcat.2014.09.024 (32) Tao, F. F.; Shan, J.; Nguyen, L.; Wang, Z.; Zhang, S.; Zhang, L.; Wu,Z.; Huang, W.; Zeng, S.; Hu, P. Nat. Commun. 2015, 6, 1.doi: 10.1038/ncomms8798 (33) Gelin, P.; Urfels, L.; Primet, M.; Tena, E. Catal. Today 2003, 83, 45.doi: 10.1016/S0920-5861(03)00215-3 Pd/Co3O4Nanoparticles Inlaid in Alkaline Al2O3Nanosheets as an Efficient Catalyst for Catalytic Oxidation of Methane LIU Jing-Wei YANG Na-Ting ZHU Yan* We report an efficient catalyst composed of ternary components prepared by inlaying Pd/Co3O4nanoparticles in alkaline Al2O3nanosheets for catalytic oxidation of methane. Pd/Co3O4inlaid in alkaline Al2O3exhibited a higher ability to break the C―H bond of methane than Pd/Co3O4supported on SiO2, ZrO2, CeO2, and acidic or neutral Al2O3. Our results show more oxygen vacancies and higher amounts of surface adsorbed oxygen on the surface of Pd/Co3O4/alkaline Al2O3than on other catalysts,which is responsible for methane activation and conversion. Further, the Pd/Co3O4/alkaline Al2O3catalyst can almost restore to its initial value in the absence of water when 5% (volume fraction) vapor water was cut off, although a decrease in activity occurred when water vapor was introduced to the reaction system.Even under a condition similar to the exhaust of a lean-burn natural gas engine, the catalytic performance of the Pd/Co3O4/alkaline Al2O3catalyst is excellent, that is, methane could be completely converted when the sample temperature in the reaction atmosphere was ramped to 400 °C. Pd/Co3O4; Alkaline Al2O3; Methane oxidation; Activity; Stability February 6, 2017; Revised: March 28, 2017; Published online: April 10, 2017. O643 10.3866/PKU.WHXB201704104 www.whxb.pku.edu.cn *Corresponding author. Email: zhuy@sari.ac.cn. The project was supported by the National Natural Science Foundation of China (21273151) and Energy Technologies Institute LLP, England.国家自然科学基金(21273151)和英国能源化学研究所研究基金资助项目 © Editorial office of Acta Physico-Chimica Sinica

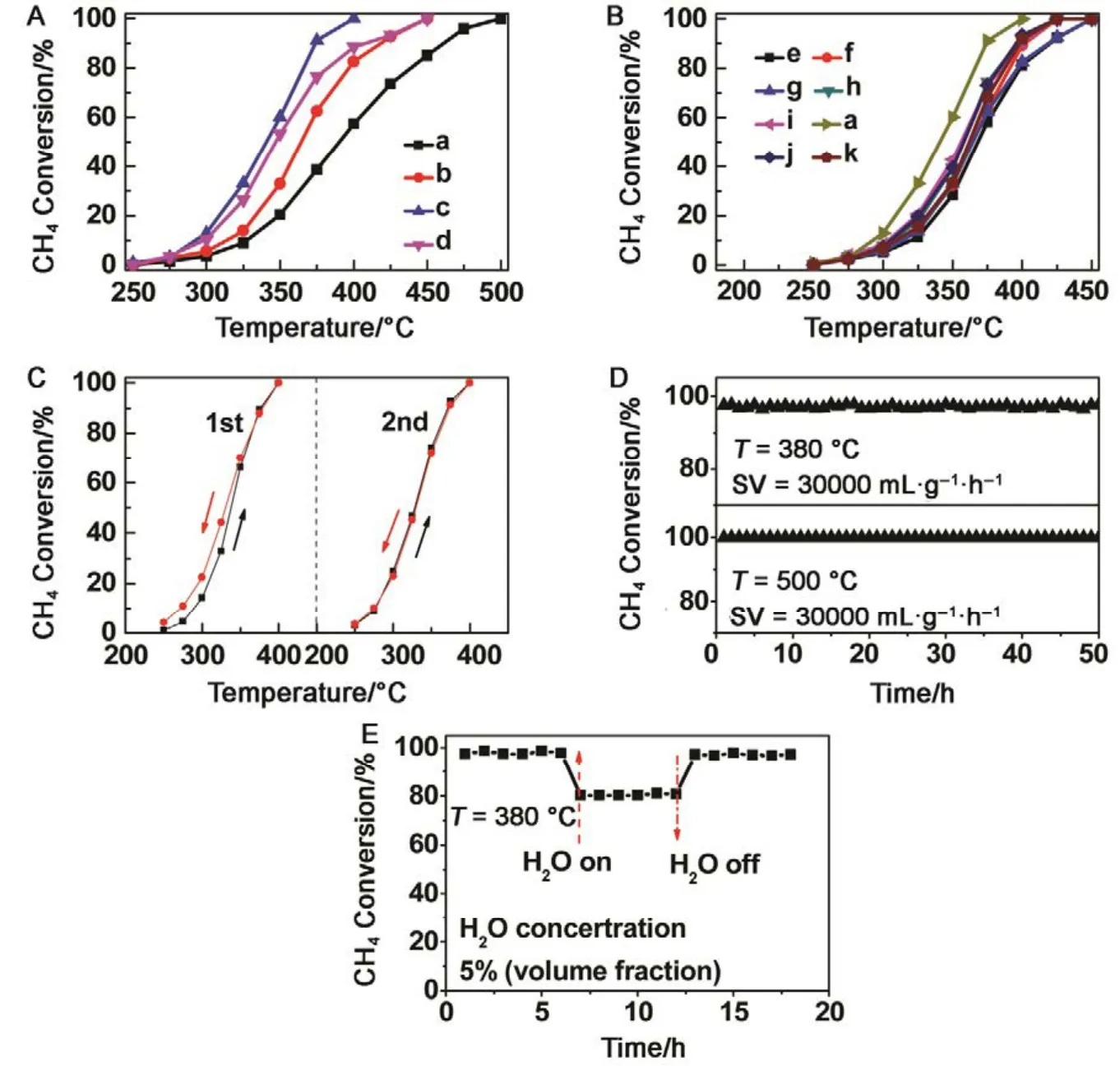

4 Conclusions
(CAS Key Laboratory of Low-Carbon Conversion Science and Engineering, Shanghai Advanced Research Institute,Chinese Academy of Sciences, Shanghai 201210, P. R. China)
猜你喜欢
中国科学院院刊(2023年2期)2023-02-27 09:55:44科学大众(2022年23期)2023-01-30 07:04:00
——李振声干旱地区农业研究(2022年3期)2022-06-09 08:45:04中山大学学报(自然科学版)(中英文)(2020年1期)2020-02-26 11:06:28红蜻蜓·低年级(2017年10期)2017-11-21 20:03:39中国组织化学与细胞化学杂志(2017年1期)2017-06-15 20:27:45浙江农业科学(2016年11期)2016-05-04 04:16:45中国科学院院刊(2016年8期)2016-03-24 09:29:45广州大学学报(自然科学版)(2015年4期)2015-12-23 11:50:10电源技术(2015年2期)2015-08-22 11:27:56
