
镍族金属团簇在催化加氢过程中的应用
2017-11-01 17:29程寒松
物理化学学报 2017年7期
韩 波 程寒松
(中国地质大学(武汉)材料科学与化学学院,可持续能源实验室,武汉 430074)
镍族金属团簇在催化加氢过程中的应用
韩 波 程寒松*
(中国地质大学(武汉)材料科学与化学学院,可持续能源实验室,武汉 430074)
贵金属纳米颗粒具有优异的催化活性,是异相催化反应中的重要角色。作为一种理想的研究模型,气相金属团簇被广泛应用于在原子和分子尺度探究催化反应的机理。在本专论中,我们将本课题组近年来关于氢气在镍族金属团簇上的解离吸附进行了回顾。首先,我们对比了不同金属团簇的结构演化规律和相对稳定性。随后,我们系统研究了H2分子在金属团簇上的解离吸附行为,揭示了不同金属对H―H键的解离能力。为了表征不同金属团簇的催化活性,我们定义了两个关键参数:氢气的解离吸附能(ΔECE)和H原子的连续脱附能(ΔEDE)。结果显示,随着H覆盖度的增大,ΔECE和ΔEDE都呈现显著的下降。由于在实际的催化反应中,氢气总是维持在一定的分压下,这就意味着催化剂金属应该总是处于较高的H覆盖度下。因此,通过处于H饱和状态下的ΔECE和ΔEDE来评估金属团簇的催化能力是合理可行的。我们发现,在饱和H吸附状态下,每一个Pt原子可以容纳4个H原子,而每一个Pd或Ni原子则只能吸附2个H原子。考虑到H原子在这些团簇上的脱附能力相当,Pt团簇相对较高的H吸附量将极大提高其在加氢过程中的催化活性。最后,我们系统研究了带电状态对Pt团簇催化性能的影响规律。结果显示,在H覆盖度较低时,H2分子的解离以及H原子的脱附过程受Pt团簇带电状态的影响较大。在饱和H吸附时,由于大量H原子的吸附,电荷的影响被平均化到每个Pt―H键上,导致ΔECE和ΔEDE都收敛到一个非常小的区域。此外,当团簇的尺寸增大时,其所带的电荷被大量的Pt原子分摊,每个Pt原子仅携带极少的电荷,使得电荷的影响已经可以忽略。
团簇;过渡金属;催化加氢;饱和氢吸附;带电状态;密度泛函理论
1 Introduction
Catalytic hydrogenation on transition metal catalysts is of great industrial interest and has been extensively used in petrochemical, agrochemical and pharmaceutical processes1.Design and development of catalysts for effective hydrogenation of olefins under mild conditions has been a subject of intensive studies in the past few decades2−4. Nickel family metals, in particular, are among the most important catalysts used in many industrial processes due to their unique catalytic activity and selectivity5−7. In a typical catalytic hydrogenation process, H2molecules initially undergo dissociation on catalyst surfaces to form metal hydride followed by H atom desorption to attack target molecules.Therefore, the dissociative adsorption energy of H2(ΔECE) and the desorption energy of H atom (ΔEDE) are the two most important chemical properties that dictate catalytic activity of a metal catalyst. In order to develop highly efficient and low-cost catalysts for hydrogenation, tremendous efforts have been made to understand the mechanisms of the catalytic processes on nickel family metal surfaces8−13. Experimentally, the ΔECEvalue on Pt(111) surface was found to be in the range between 0.70 and 0.83 eV14,15. Theoretically, the calculated ΔECEon the Pt(111) surface falls in the range of 0.80 to 1.54 eV,significantly higher than the experimental values16−18. For Pd(111) surface, the ΔECEwas found to be much lower than the value on Pt(111) and the diffusion energies and dissociation barriers of H2on Pd(111) were correlated to the H2coverage on the substrate19−21.
Since nano-sized particles are chemically more active than bulk-sized catalyst of the same element, these metal catalysts are often dispersed in a form of nanoparticles or clusters on various support materials22−30. Generally, compared with the bulk form, small size metal clusters exhibit quite different physicochemical properties. The catalyst particles have more edges and corners that are responsible for catalytic reactions.Liu et al.31−33reported a significantly enhanced H2chemisorption energy of 1.36 eV on a NaY zeolite supported Pt13cluster34. Similar results have also been reported byOkamoto via DFT calculations35,36. Besides the relatively high adsorption energies of H2on transition metal clusters (TMCs),the desorption energies of atomic hydrogen adsorbed on the TMCs were also found to be much higher than the value on crystal surfaces. For instance, the H desorption on Pt(111)surface was found to be 2.60−2.65 eV37−39. However, the desorption energy was found to increase substantially to 2.9−4.5 eV when the H atom is adsorbed on a Pt13cluster40.


In addition to the size effect of catalyst particles, H2adsorption and H desorption properties are also dependent on surface coverage, which profoundly influences the catalytic activity of TMCs. Indeed, there is a significant variation on catalytic performance of metal clusters at different H loadings.Generally, at low surface coverage, H2dissociative chemisorption produces a strong adsorption energy, which declines gradually as coverage increases13. In a practical hydrogenation process, a partial pressure of hydrogen is always maintained. Therefore, surfaces of a catalyst are nearly fully saturated by the dissociated H atoms. Much attention has been paid to catalytic performance at low H coverage, relatively scarce theoretical studies on catalysis at high H coverage on TMCs have been conducted. On the other hand, a wide variety of gas phase neutral, anionic, and cationic metal clusters has been produced using techniques such as pulsed laser ablation41−43and time-of flight apparatus of supersonic beams via electron attachment or ionization44−47. It was found that there are significant differences in physicochemical properties between neutral and charged clusters. A recent combined theoretical and experimental study48on hydrogen dissociative chemisorption on small platinum clusters further showed that the number of hydrogen molecules adsorbed dissociatively on a cationic Pt cluster is significantly smaller than on a neutral cluster of the same size13,49,50.
In this paper, we present our recent first principles studies on H2dissociative chemisorption on a series of nickel family metal clusters Mn(M = Ni, Pd, Pt; n = 2−9, 13) to address their catalytic performances. The influences of H surface coverage,metal type, and charge state of clusters on catalytic hydrogenation are systematically discussed. The results are also validated by available experimental data. The purpose of the present report is to provide useful insight into the atomistic details of the chemical processes from the electronic structure calculations.
2 Gas phase TMCs
For nickel family clusters, the stable structure and growth pathway from small cluster to bulk could undergo three typical patterns, including the close-packed pattern, the icosahedral pattern and the fcc-like pattern8,51,52. Take a Pd cluster as an example. The optimized structures and the associated binding energies of Pdnclusters (n = 2−15) are summarized in Fig.1,where the structures of the isomers are also included. The Pd clusters adopt a close-packed planner configuration before tetramer, after which a 3-dimensional (3D) configuration is energetically preferred. From Pd4to Pd7, the clusters exhibit bipyramid configurations with elongated Pd-Pd bond distances and higher binding energies. A previous DFT study based on a tight-binding model53suggested that the most stable structure of Pd7is planar (Pd7-c), which is much less favorable than the bipyramid configuration in our calculation. From Pd8, only slight deviation on the binding energies between different isomers is observed, suggesting that these close-packed isomers may coexist thermodynamically. Li et al.54suggested that the Pd7and Pd8should adopt a more symmetric configuration (i.e.Pd7-b and Pd8-d). However, our results indicate that the close-packed structure is energetically more stable. In particular, Pd13-b exhibits a typical icosahedral configuration with the calculated binding energy of 2.337 eV, nearly degenerated with that of Pd13-a, which adopts a close-packed pattern.
A similar growth path for Ptnclusters to what is observed for Pdnclusters is shown in Fig.2(a), where the associated binding energy and geometric parameters are also included.Interestingly, the Pt6prefers a 2-dimensional (2D) planar configuration, while Pt4and Pt5adopt a 3D close-packed pattern, in line with the results reported by Kadıoglu et al.55. In addition to the 2D equilateral triangular structure, we also obtained an octahedral structure for Pt6whose average binding energy of 2.886 eV is only slightly smaller than that of the equilateral structure (2.941 eV). It is possible that both geometries may exist for this cluster. For Ni clusters, the calculated lowest energy cluster of a given size exhibits similar structural characteristics to the same size of Pt and Pd clusters and the Ni clusters undergo a similar triangular growth pathway(Fig.2(b)).
To understand the relative stability of the clusters, the structures and binding energies of Nin, Pdnand Ptnare systematically compared, as shown in Fig.3. We note that the Ni―Ni bond lengths fall into a narrow range between 2.35 and 2.45 Å (1 Å = 10−10m), which is relatively short due to the small size of nickel atom. However, the Pd clusters exhibit the longest bond lengths, although its atomic size is smaller than Pt.The long Pd−Pd distances give rise to the more relaxed packing configurations of Pd clusters, leading to weak binding strengths.Indeed, the calculated binding energies of the Pd clusters are much lower than the values of the corresponding Pt clusters which display the strongest bind strengths. The average binding energy increases with the cluster size monotonically. For the same cluster size, the binding strength of the Ni cluster is similar to that of the Pd cluster but smaller than that of the Pt cluster. The relative binding strength of small clusters shown in Fig.3 is consistent with the trend of the cohesive energies of the bulk Ni, Pd and Pt solids.
3 H2 adsorption and dissociation on TMCs

Fig.1 Optimized structures for Pdn clusters (n = 2−15)52.Reprinted with permission from Wiley Periodicals, Inc.
We next focus on the sequential H2dissociative chemisorption on the lowest energy TMCs. Fig.4 displays the H2dissociative process on the Pt2cluster13. The H2molecule approaches the Pt2dimer from one end of the dimer axis with the Pt atom vertically toward the center of mass of H2to form a weakly bonded species (R). Subsequently, the R undergoes a transition state (TS1) with a small energy barrier of 0.05 eV that leads to decomposition of H2. The dissociative chemisorption leads to the formation of a T-shaped adsorption structure (P1) with the calculated exothermic reaction energy of−0.26 eV. The adsorbed H atom may diffuse to the other side of the dimer (P2) to further stabilize the adsorption structure with an energy barrier of 0.13 eV (TS2). Overall, the calculated chemisorption energy is 1.78 eV. The strong chemisorption arises from the orbital overlaps between the 1s orbital of H atoms and the 5d orbitals of Pt atoms, resulting in charge transfer from Pt 5d to H 1s. For large cluster (Pt13), similar reaction pathway was identified with the calculated activation barriers of 0.06 and 0.34 eV for TS1 and TS2, respectively.Recently, Ignatov et al.56reported the adsorption behavior of H2on a Pt24subnanoparticle, where the dissociation of H2and migration of H atom were also found kinetically favorable with moderate activation barriers (less than 0.2 eV). In fact, for Ni8and Pd12clusters (Fig.5), the dissociation process is essentially barrierless, similar to the situation of H2 dissociation on Pt clusters57. The calculated H2adsorption energy on a Ni6cluster is 1.31 eV, slightly higher than the value on Pd6 (1.16 eV) but considerably lower than on Pt6 (1.61 eV). The H migration on Ni6requires only 0.19 eV activation energy, which is slightly higher than that on Pd6(0.11 eV) but slightly lower than on Pt6(0.23 eV). The modest difference in the calculated H migration barriers among these clusters correlates well with the relative stability of H2adsorption on the clusters. Our calculations suggest that the H2dissociative chemisorption requires very little activation energy and the adsorbed H atoms are highly mobile.

Fig.2 Optimized configurations of (a) Ptn clusters and (b) Nin clusters (n = 2−9, 13).
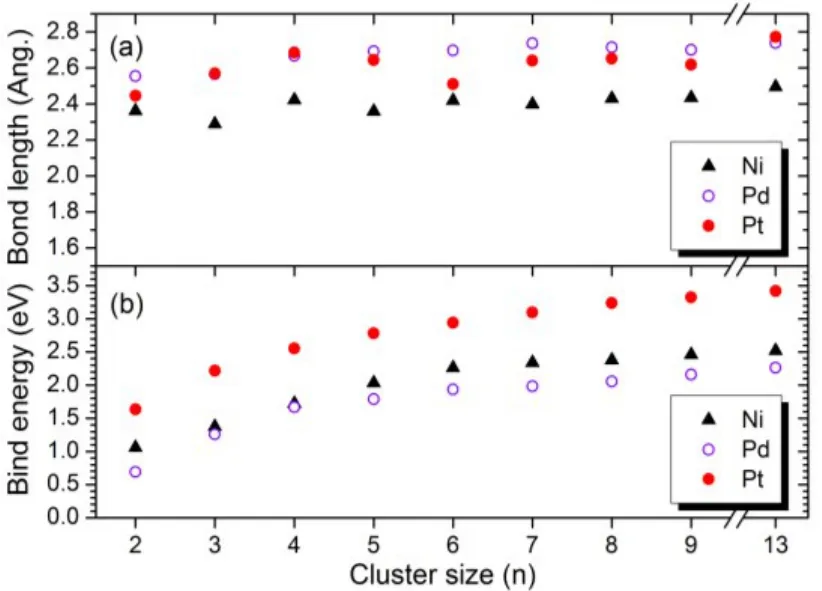
Fig.3 Comparison of the calculated bond length and binding energies of small Ni, Pd and Pt clusters8.
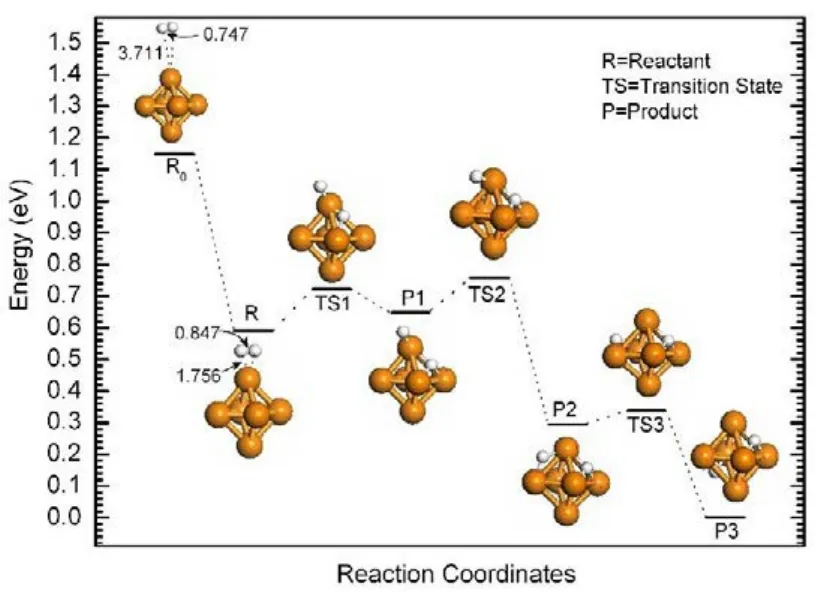
Fig.5 Energy diagram of H2 dissociation and diffusion onthe Pd6 octahedral cluster.
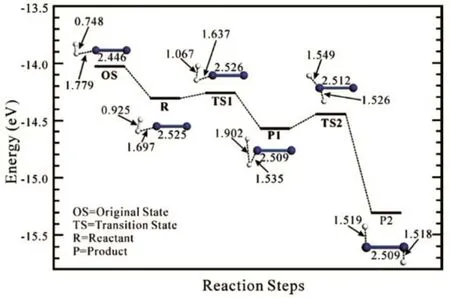
Fig.4 Energy diagram of H2 dissociative chemisorption and diffusion on the Pt2 cluster.
In general, H2dissociative chemisorption energy (ΔECE), H sequential desorption energy (ΔEDE) and H capacity are the key factors that dictate catalytic performance of TMCs. To understand the coverage effect on the values of ΔECEand ΔEDE,sequential H2chemisorption on Pt clusters was simulated. For Pt2, the calculated ΔECEand ΔEDEare shown in Fig.6, (a1) and(b1), respectively. The Pt2cluster exhibits a relatively high H capacity up to 10 H atoms, after which additional two H atoms are recombined to form a H2molecule. Both ΔECEand ΔEDEdecrease as the number of H atoms on the cluster increases.Upon saturation, the calculated chemisorption energy is 1.1 eV,about 0.3−0.4 eV higher than the experimental value for H2on the Pt(111) surface16. It is interesting to note that the value of H2dissociation energy ΔECEdrops substantially from 1.78 eV at low coverage to 1.1 eV at full saturation, while the threshold H-desorption energy ΔEDEat full coverage is about 0.6 eV lower than its value at low coverage. The amount of the calculated Hirshfeld charge transferred from Pt2to the H atoms(ΔQ) is shown in Fig.6(c1), which declines with the number of H atoms. For the Pt3trimer, the calculated ΔECE, ΔEDE, and ΔQ exhibit almost the same trends as for the Pt2dimer. The Pt3cluster can accommodate up to 12 H atoms. Substantial structural expansion as well as distortion of the triangular geometry of the cluster is observed upon hydrogen uptake.
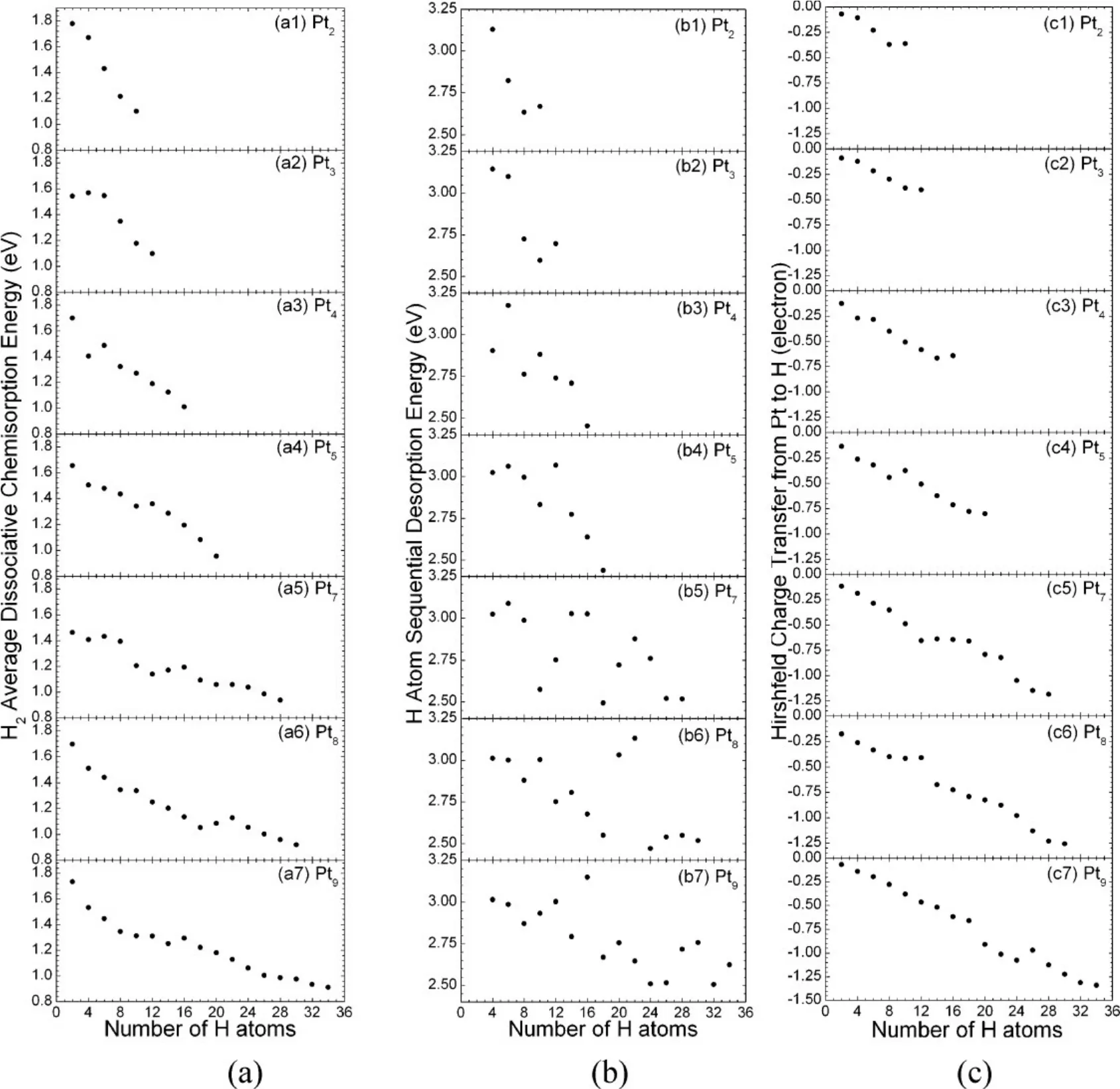
Fig.6 Calculated (a) ΔECE, (b) ΔEDE, and the loss of the Hirshfeld charges of Pt clusters with respect to H coverage13.
To show the H2sequential adsorption on Pt clusters, the Pt4clusters with various H coverages were selected as a model system, as shown in Fig.7(a). The bare Pt4cluster adopts a tetrahedral configuration. Upon H loading, the cluster is distorted to a certain extent. Full saturation is realized at H capacity of 16. In contrast, loading 18 H atoms on the cluster would result in recombination of 2 H atoms to form H2molecule during ab initio MD simulations. It was observed from the MD trajectories that two H atoms are readily squeezed out from the cluster, forming a H2molecule that is subsequently bonded to the cluster via a weak interaction force.The calculated H―H bond distance distribution for Pt4H16and Pt4H18are shown in Fig.7(b). The smallest H-H distance for Pt4H16is around 2.1 Å, indicating complete dissociation of H2molecules. In contrast, a peak around 0.75 Å corresponding to the formation of H2molecule was observed for Pt4H18. Once again, more charge flows from the Pt4cluster to H atoms as the H coverage increases (Fig.6(c2)). or larger Ptn(n > 4) clusters,the calculated ΔECE, ΔEDE, and ΔQ exhibit similar features to what were found for smaller clusters (Fig.6). The general trend of these quantities is that they decline with the H coverage.However, while both ΔECEand ΔQ decrease monotonically with the number of H atoms, some fluctuation of ΔEDEis observed. This is because H atoms first saturate the energetically most favorable sites. As these sites become filled,their stability decreases and the vacant sites begin to fill. The fluctuation of ΔEDEis due to the difference in chemisorption energies at different adsorption sites near the saturation boundary. The full H saturation is determined by MD simulations with the same procedure as Pt4. We note that in all cases, the H2dissociative chemisorption energy fluctuates in the range between 0.9 and 1.1 eV at the full saturation, slightly higher than the experimental value for the H2dissociative chemisorption on the Pt(111) surface at zero coverage by 0.2−0.3 eV. Similarly, the calculated threshold H sequential desorption energy varies in a narrow range of 2.45−2.60 eV,slightly smaller than the value for an isolated H atom on the Pt(111) surface reported in a previous study by Papoian and coworkers16. Fig.8 summarizes the calculated ΔECEand ΔEDEat low and high H coverages. Both ΔECEand ΔEDEare significantly reduced at high H coverage. For ΔECE, a large energy gap of 0.8 eV is observed between the low and high H coverages. Meanwhile, an energy gap of 0.5 eV is emerged in the calculated ΔEDE. Our results suggest that both the H2dissociative chemisorption energy and H desorption energy on Pt clusters are strongly coverage dependent.

Fig.7 (a) Fully optimized structures of H2 sequential dissociative chemisorption on a Pt4 cluster; (b) calculated H-H distance distribution for Pt4H16 and Pt4H18, respectively13.
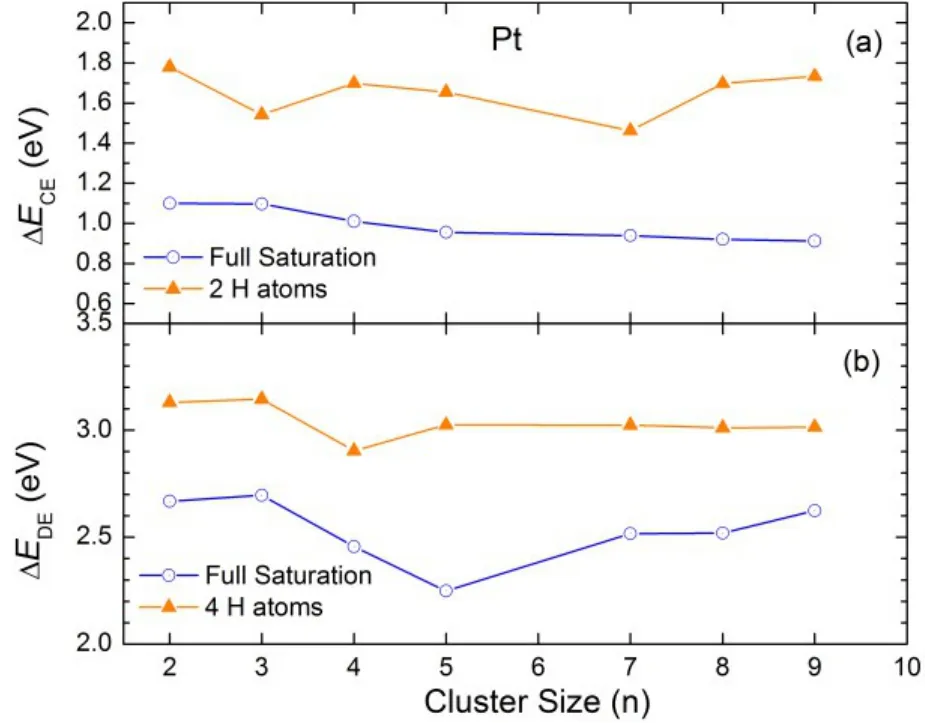
Fig.8 Comparison of the calculated (a) ΔECE and (b) ΔEDE on Pt clusters at low and full H coverages13.
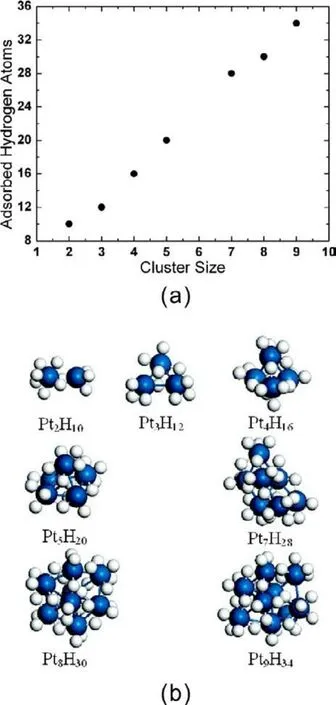
Fig.9 (a) Number of H atoms adsorbed on Ptn cluster at fully H coverage; (b) optimized structures of Pt hydrides13.
To identify the H capacity of Ptnclusters at various size, the number of H atoms versus the size of clusters at full saturation is shown in Fig.9, where the optimized structures of Pt hydrides are also included. It shows that the number of H atoms increases almost linearly with the size of clusters. We expect such a quasi-linear relationship would not hold for larger clusters since only the surface Pt atoms are accessible for H2.Nevertheless, the catalytic efficiency of small clusters for H2dissociative chemisorption is still remarkable. The H-capacity could be revealed by the H : Metal ratio, which approximately gives an H:Pt ratio of 4 based on our results.
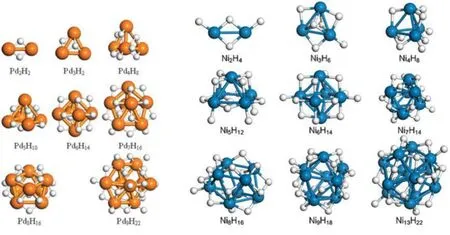
Fig.10 Optimized structures of Pdn and Nin clusters at full H coverage8,12.
For Pd and Ni clusters, similar H2dissociation processes were simulated. Fig.10 shows the fully saturated hydrides of the selected Ninand Pdnclusters. We note that, H atoms prefer the 2-fold edge sites on both Pd and Ni clusters. In contrast, the 1-fold on-top site is energetically more stable for Pt cluster(Fig.9(a)). Although most of the H atoms are located at the edge sites, some of the on-top sites are also populated for large clusters and at high H loadings, at which the edge sites become fully occupied. The H capacities of the selected Pt, Pd and Ni clusters are summarized in Fig.11(c). The results indicate that at a given size the maximum hydrogen capacity of a Ni cluster is virtually identical to that of the Pd cluster, despite the fact that the size of Pd cluster is considerably larger than Ni with the same metal atoms. Interestingly, the capacity of small Pd and Ni clusters to absorb H atoms is essentially half of what was found for Pt clusters, which not only have higher binding strengths with H atoms but also allow H atoms to reside at the on-top sites.
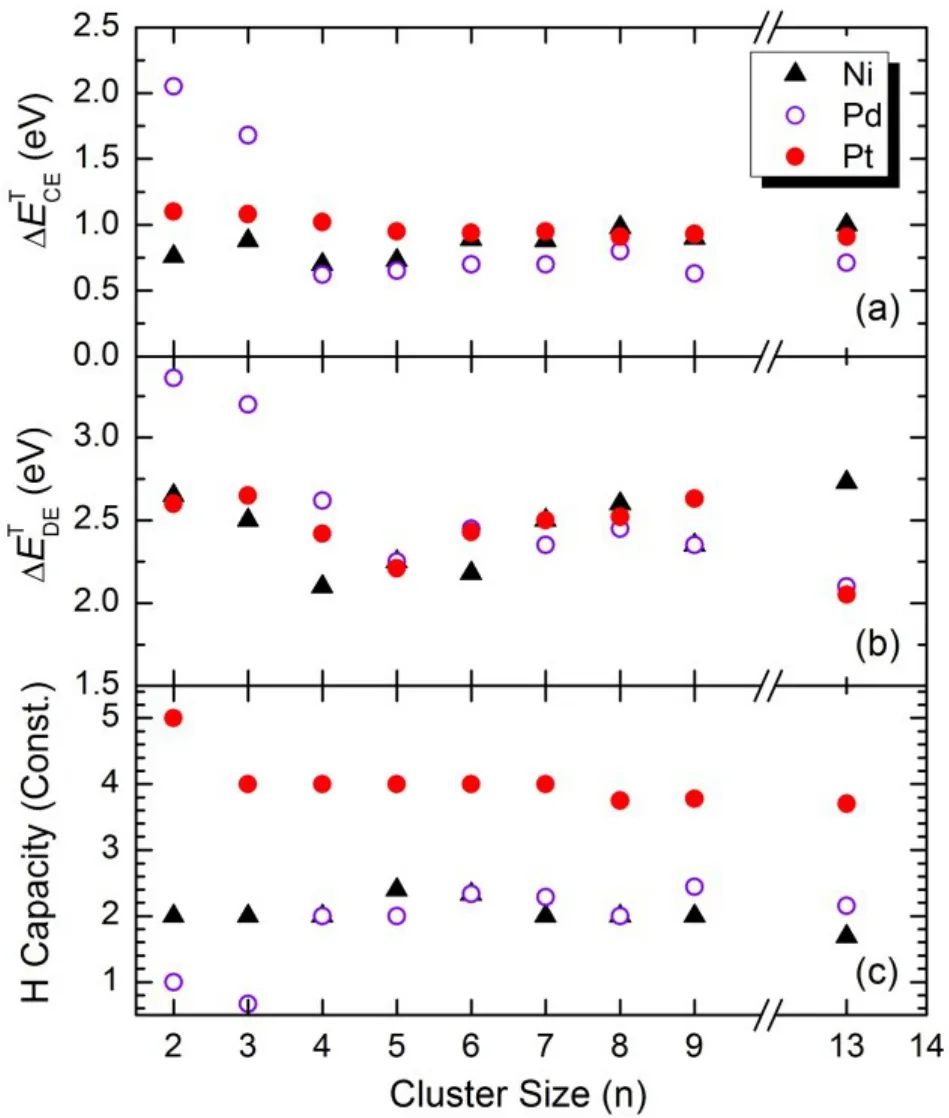
Fig.11 Comparison on (a) the threshold of H2 dissociative chemisorption energy, (b) the threshold of H desorption energy and(c) the maximum H-capacity of small Ni, Pd and Pt clusters8.
Fig.11(a) compares the calculated average H2dissociative chemisorption energies (ΔE) of Nin, Pdnand Ptnclusters at full saturation. Remarkably, they all vary in a small energy range between 0.6 and 1.0 eV regardless of the cluster size except for Pd2and Pd3, which exhibit considerably higher average H2dissociative chemisorption energies. The results imply that clusters of the three metal elements behave similarly in terms of dissociating H2molecules at full H coverage. The size-independence is largely due to the fact that the chemical bonding of the clusters evolves from metallic to covalent as H atoms are sequentially loaded up onto the clusters. As a consequence, the chemical properties become localized and thus do not depend strongly on cluster size. We note that at typical catalytic hydrogenation conditions the metal catalysts are fully covered by H atoms since a given hydrogen pressure is always maintained. For a given catalyst quantity, smaller catalyst particles offer higher surface area exposed to the gas species. However, our results suggest that H2dissociative chemisorption energies do not change significantly with particle size. This makes computational modeling of surface catalysis convenient because one could select a relatively small size cluster to represent catalyst particles. Similarly, we calculated H atom desorption energies (ΔE) at full H coverage of the metal clusters and the results are displayed in Fig.11(b). The desorption energy at full H coverage is an important physical quantity because it can serve as an indicator of catalytic activity. A small desorption energy indicates higher activity. For small Ninclusters, the H desorption energy varies in a range between 2.08 to 2.73 eV. The highest desorption energy is observed for Ni13, sharply higher than the values of Pd13and Pt13. The results suggest that it is more difficult to pull H atoms out of the Ni catalyst than out of Pd and Pt catalysts.Fig.11(c) displays the calculated average H capacities of Nin,Pdnand Ptnclusters. Essentially, each Pt atom is capable of adsorbing 4 H atoms, while each Ni or Pd atom can only accommodate 2 H atoms. Pelzer et al.58reported a much higher H2uptake on small Pdnclusters (n < 5), where each Pd atom could adsorb more than 4 H atoms. However, instead of dissociative chemisorption, most H2molecules were only physically attached on Pd atoms in there simulations.Considering the similar H desorption energies on the Pdnand Ptnclusters, the relatively high H capacity of Ptngives rise to a superior reaction rate since more active H atoms are provided with Pt catalyst particles in hydrogenation processes.
4 Charge effect
We next consider the charge effect on the catalytic activity of TMCs. It was reported that, charged clusters could exhibit significant differences in physicochemical properties, leading to substantial variation on catalytic performances59−62. Indeed,these experimental observations have raised a fundamental question on whether a metal cluster in its charged state would maintain similar catalytic properties to the cluster in its charge-neutral form and, more profoundly, on whether clusters could indeed serve as a valid model for catalyst particles regardless of their charge states. Generally, metal catalysts used in practice are charge neutral, while metal clusters produced in spectroscopic experiments could be either cationic or anionic.Moreover, the charge state of a metal catalyst on a supporting material may also be affected by the interaction between the catalyst and the substrate via electron transfer, which in turn may influence the activity of the catalyst28,63,64. Here, DFT calculations on selected Pt clusters (Pt2, Pt4, Pt6and Pt8) were carried out to determine the impact of charges on the reactivity of catalysts through a systematic analysis of the key catalytic properties for hydrogenation, including ΔECE, ΔEDEand H-capacity.
We first chose neutral Pt4, cationic Pt4+and anionic Pt4−clusters as probes to identify the differences upon sequential H2dissociative chemisorption. A tetrahedral configuration with negligible structural deformation was observed for all bare Pt4clusters, regardless of their charge state. The average Pt−Pt bond distance is gradually elongated from 2.654 to 2.728 Å with respect to the enrichment of the electron density on Pt4.The calculated electron density difference of the neutral and charged states of Pt4and the d-band center of Pt clusters with various sizes and charges are shown in Fig.12. As expected, Pt4+is most electron deficient, while Pt4−holds the highest electron density. The charge distribution on the clusters profoundly influences their capability on catalytic hydrogenation, in which the activity is highly correlated to the electron transfer from Pt to H2. Fig.12(b) reveals that the d-band center decreases gradually with the cluster size. In general, for a given cluster size, an anionic cluster exhibits a higher d-band center, while a cationic cluster displays a lower d-band center. However, the d-band center gradually converges as the cluster size increases,regardless of the charges on the clusters. Our results also suggest that the d-band center of large clusters becomes insensitive to the charge states.
To understand the influence of the charge state on catalytic dissociation of H2, minimal energy pathway calculations of H2dissociative chemisorption were carried out on the Pt4+, Pt4and Pt4-clusters, respectively, and the results are shown in Fig.13.The associated energy profiles for the Pt4+, Pt4and Pt4−clusters are compared in Fig.14. For the neutral Pt4, H2dissociation exhibits a reaction pathway similar to that of Pt2(Fig.4). Here,the H2molecule adsorbed on the atop-site of Pt4undergoes spontaneous dissociation, leading to two atomic H species adsorbed on the same Pt atom. Considering the high mobility of H atoms on TMCs, one H atom can easily diffuse to the bridge-site (P1) with a moderate activation barrier of 0.466 eV to further stabilize the hydride. The strong chemisorption is attributed to the large orbital overlap between the 1s orbital of H and the 5d orbitals of the Pt atoms, resulting in electron transfer from Pt to H. As a result, the electron rich Pt4−cluster exhibits a higher H2dissociative chemisorption energy. Indeed,our results show that H2dissociation on the electron rich anionic Pt4−cluster follows a similar reaction pathway(Fig.13(c)) but with a stronger Pt-H interaction.
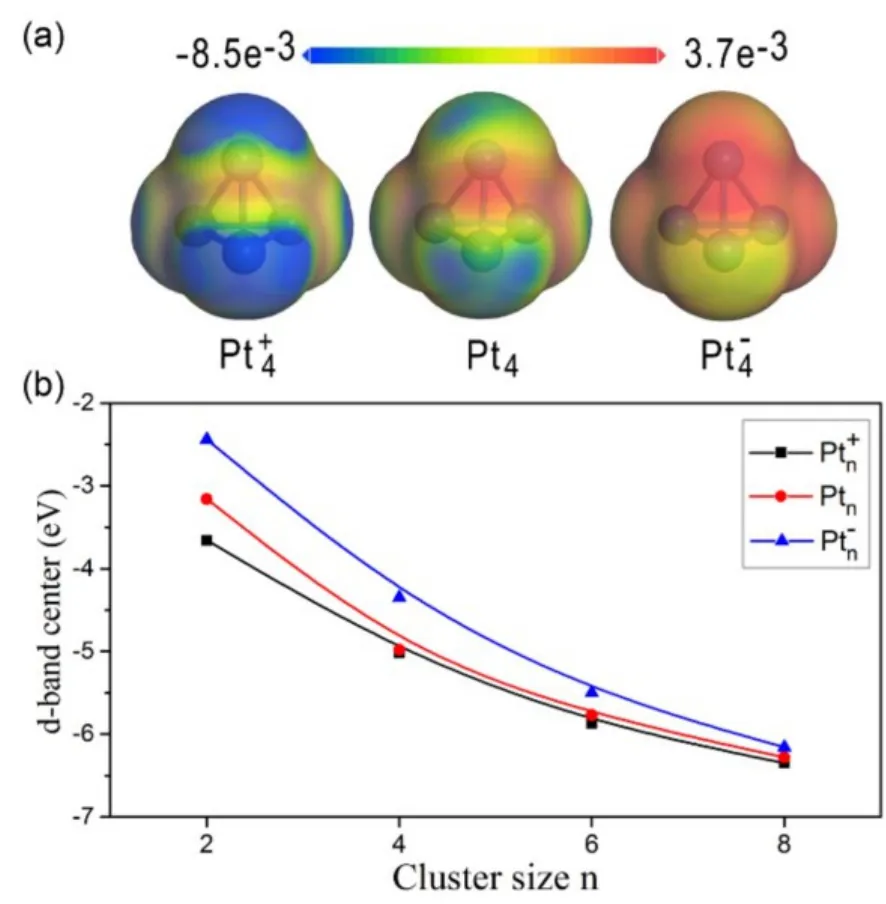
Fig.12 (a) Calculated electron density difference of Pt4+,Pt4 and Pt4−; (b) d-band center of the clusters with various sizes and charges61.
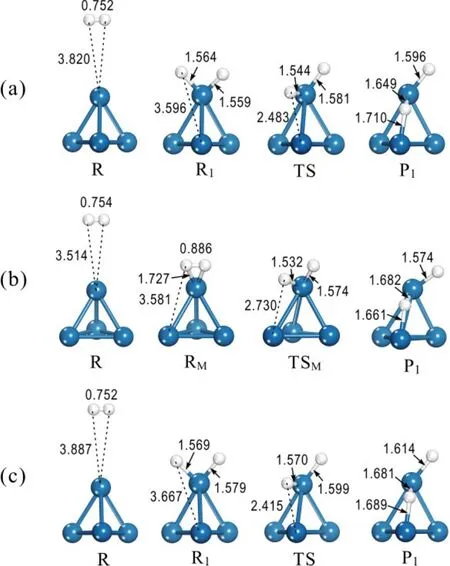
Fig.13 Reaction pathway of H2 activated by (a) the Pt4,(b) the Ptand (c) the Pt cluster.

Fig.14 Calculated energy diagram of H2 molecule dissociative chemisorption on the Pt4 clusters61.
Typically, dissociation of H2is catalyzed by electron migration from d-orbitals of the metal to the anti-bonding π*-orbital of H2, as have been observed for the Pt4and Pt4−clusters. However, for the electron deficient cationic Pt4+cluster, a small electron transfer from the bonding σ-orbital of H2to the metal cluster is observed, resulting in a considerably weaker force to break up the H―H bond. In fact, the H2molecule may be molecularly anchored on Pt4+upon contact with the cluster (Fig.13(b)). The molecularly bonded H2is also chemisorbed and slightly activated with a H―H bond distance of 0.886 Å. A similar phenomenon of molecular H2adsorption on a cationic Ni4+cluster has been observed previously in a vibrational spectroscopic (IR-MPD) study by Swart and co-workers43. In the present case, the calculated adsorption energy on the cationic Pt4+cluster (−0.817 eV) is only half of the value on the neutral Pt4cluster (−1.650 eV). An energy barrier of 0.310 eV is required for dissociation of the molecularly adsorbed H2species (RM) into two H atoms, which subsequently migrate to the most stable chemisorption sites on the Pt4+cluster (P1) to further enhance the dissociative chemisorption of H2by 0.284 eV. Our results indicate that H migration on Pt4+is easier than that on Pt4due to the weaker binding strength on the cationic cluster. Charge distribution analysis indicates that the H atoms gain only a small fraction of electron (0.046e) from the Pt4+cluster at the most stable binding sites (P1). In contrast, the excess electron on Pt4−gives rise to significantly enhanced electron transfer from the cluster to the H atoms (0.242e at P1), which is almost twice the value found for the neutral case. As a consequence, the H2dissociation process on Pt4−becomes highly exothermic with the calculated chemisorption energy of −2.117 eV. Not surprisingly, the diffusion of H atoms on the anionic cluster is somewhat restricted due to the strong Pt-H interactions. An activation barrier of 0.554 eV, which is slightly larger than on Pt4and Pt4+,is required to move a H atom to the most stable site (P1).

Fig.15 Calculated H-H distance distribution function g(r) of the fully H saturated structures, obtained by tabulating all the H−H distances at each step of the MD trajectories61.
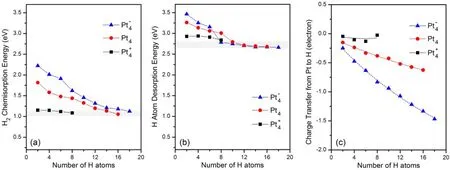
Fig.16 Calculated H2 average dissociative chemisorption energy (a), H sequential desorption energy (b), and the loss of the Hirshfeld charges (c)61.
The above results indicate that H2chemisorption is very sensitive to the charge state of a cluster at low H2loadings.While these results are consistent with experiments on small clusters, they provide limited insight into the reactivity of a realistic catalyst, in which the metal surface is often saturated with hydrogen atoms/molecules. To address the effect of H coverage on Pt clusters with different charges, the sequential dissociative chemisorption of H2molecules on the clusters was examined until full saturation, where all H2molecules were completely dissociated into H atoms and adsorbed on the clusters (Fig.15). The maximum H atom loadings on the Pt4+,Pt4and Pt4−clusters were evaluated to be 8, 16, and 18 H atoms,respectively. As mentioned in Section 3, the saturation limit for each of these clusters was tested by MD simulations, during which loading two more H atoms onto the saturated clusters yields a H2molecule (peak at ca 0.75 Å) weakly bonded to the clusters. These results indicate that the charge state of a Pt cluster significantly affects the H-capacity of the cluster, which increases with the number of electrons for a given cluster size.In particular, the H-capacity of the Pt4+cluster is only half of the value of the neutral cluster, in agreement with the results reported by Kerpal and co-workers65. Since the breaking of the H2bond is the main process during a catalytic hydrogenation reaction, the Pt4−cluster would provide the highest catalytic reactivity, while the Pt4+cluster with the significantly reduced H-capacity would yield the lowest activity.
The high sensitivity of the H-capacity to the charge state indicates that a Pt4cluster is too small to effectively represent a nanoparticle catalyst. In order to gain additional insight into the variation of the H-capacity for different charge states, we calculated the values of ΔECE, ΔEDEand ΔQ as a function of H coverage for the Pt4+, Pt4and Pt4-clusters. The results are shown in Fig.16. Clearly, ΔECEdecreases monotonically as H coverage increases and thus, thermodynamically, dissociation of H2becomes increasingly more difficult. Despite the distinctive difference in H-capacities of these clusters, the calculated average chemisorption energies at full H saturation in all cases fall into a narrow range (ca 1.05−1.15 eV). These results suggest that all 3 clusters to some extent hold the same binding strength toward H atoms at high H loadings. Likewise,all the calculated ΔEDEvalues at full H coverage fall within a narrow range (ca 2.65−2.85 eV) despite the large energy variation observed at low H loadings. Although the chemisorption energies and H desorption energies on different charged Pt clusters are similar at full H saturation, the charge transfer shown in Fig.16(c) is quite different. This difference is responsible for the significant variation in H-capacity for different charge states of the Pt4cluster.
To understand the effect of charge states on the H-capacity for different cluster sizes, we further calculated the saturated hydrogen complexes of Pt2, Pt6and Pt8clusters with different charge states at full saturation. The H/Pt ratio vs clusters size at full saturation is shown in Fig.17. In general, we found that more H atoms are loaded onto an anionic Ptn−cluster and fewer H atoms are attached to a cationic Ptn+cluster. For anionic Ptn−cluster, the H/Pt ratio decreases from 5.00 to 4.25 as the clusters size increases from 2 to 8. For cationic Ptn+clusters,the H/Pt ratio initially decreases with cluster size before increasing from 2.00 to 3.75 as the clusters size increases from 4 to 8. The H/Pt ratio gradually converges to 4.00 as the size of clusters increases, independent of the charges on the clusters.Therefore, the charge effect would be significantly diminished for large size clusters with high H loadings. It is in this limit(H/Pt ratio equal to 4 at full H saturation) that a small cluster may be used to model catalyst particles. In this case, the cationic or anionic charge can be readily dispersed and a sufficient number of Pt―H bonds is involved to average the interaction between the cluster and H atoms. This phenomenon is consistent with the experiments for cationic Pt and Ni clusters, where the reactivity of metal clusters was observed to increase significantly as the cluster size increases at a subnanoscale43,65. The reason for the increased catalytic reactivity is mainly attributed to the increased H-capacity for large cationic clusters in which the charge effect is effectively minimized. Our results demonstrate that H-capacities on charged clusters become similar for large size clusters, and the reactivity of a fully saturated cluster is no longer sensitive to its charge state if the cluster size exceeds a certain number of atoms (e.g. ~8 Pt atoms).

Fig.17 H/Pt ratio versus cluster size at full saturation61.
5 Conclusions
Design and development of catalysts for effective hydrogenation of olefins under mild conditions has been a subject of intense studies in the past few decades. Nickel family metals have been used as the most important catalysts in many industrial heterogeneous catalytic processes due to their unique catalytic activity and high selectivity. In this review, we summarized our recent first principles studies on H2dissociative chemisorption on a series of nickel family metal clusters Mn(M = Ni, Pd, Pt; n = 2−9, 13) to address their catalytic performances. We first compared the growth pathways of Ni, Pd and Pt clusters. It was found that the metal clusters prefer to grow in a 3D close-packing growth pattern.The Pd clusters exhibit the longest bond lengths, although its atomic size is smaller than Pt. The average binding energy of the metal clusters increases with the cluster size monotonically.For a given cluster size, the binding strength of a Ni cluster is similar to that of the Pd cluster but smaller than that of the Pt cluster. Subsequently, H2dissociative chemisorption on metal clusters was systematically addressed. It was found that H2dissociation is almost barrierless on the Ni, Pd and Pt clusters.A moderate activation energy is required for H atom migration to the most stable adsorption site, indicating that the H atoms are highly mobile on metal clusters. Two key energy parameters, the H2dissociative chemisorption energy (ΔECE)and H sequential desorption energy (ΔEDE), were defined as the probe to characterize the catalytic activity of metal clusters.Both ΔECEand ΔEDEdecrease significantly as the number of H atoms on the cluster increases. Upon saturation, large energy gaps were observed between low and high H coverages for both ΔECEand ΔEDE. However, the ΔECEvalue of the Ni, Pd and Pt clusters at full H saturation vary in a small energy range between 0.6 and 1.0 eV regardless of the cluster size. We found that it is more difficult to pull H atoms out from the Ni clusters than from the Pd and Pt clusters. At full H coverage, each Pt atom is essentially capable of adsorbing 4 H atoms, while each Ni or Pd atom can only accommodate 2 H atoms. Considering the similar H desorption energy on the Pt and Pd clusters, the relatively high H capacity of Pt would lead to a superior reaction rate since more active H atoms are available for a hydrogenation process. In addition, charge sensitivity of the key energy parameters for several selected Pt clusters was also systematically evaluated. The results show that the dissociation of H2and H-capacity are strongly correlated to the charge state of the Pt clusters at low H coverage. However, at high H-capacity, both ΔECEand ΔEDEfall into a narrow energy range, suggesting that the charge effect can be readily diminished and a sufficient number of Pt―H bonds is involved to average the interaction between cluster and H atoms for large size clusters. As a result, the H-capacities on different charged clusters become similar if the cluster size becomes sufficiently large, and the reactivity of the fully saturated cluster is no longer sensitive to its charge state. The present review provides useful insight into the reactivity of small nickel metal family clusters for catalytic hydrogenation.
Supporting Information: available free of charge via the internet at http://www.whxb.pku.edu.cn.
(1) Chen, A.; Holt-Hindle, P. Chem. Rev. 2010, 110, 3767.doi: 10.1021/cr9003902
(3) Biffis, A.; Zecca, M.; Basato, M. J. Mol. Catal. A: Chem. 2001, 173,249. doi: 10.1016/S1381-1169(01)00153-4
(4) Lang, S. M.; Bernhardt, T. M. Phys. Chem. Chem. Phys. 2012, 14,9255. doi: 10.1039/c2cp40660h
(5) Tao, F.; Grass, M. E.; Zhang, Y.; Butcher, D. R.; Renzas, J. R.; Liu,Z.; Chung, J. Y.; Mun, B. S.; Salmeron, M.; Somorjai, G. A. Science 2008, 322, 932. doi: 10.1126/science.1164170
(6) Lim, B.; Jiang, M.; Camargo, P. H. C.; Cho, E. C.; Tao, J.; Lu, X.;Zhu, Y.; Xia, Y. Science 2009, 324, 1302.doi: 10.1126/science.1170377
(7) Imaoka, T.; Kitazawa, H.; Chun, W. J.; Omura, S.; Albrecht, K.;Yamamoto, K. J. Am. Chem. Soc. 2013, 135, 13089.doi: 10.1021/ja405922m
(8) Zhou, C. G.; Yao, S. J.; Zhang, Q. F.; Wu, J. P.; Yang, M.; Forrey, R.C.; Cheng, H. S. J. Mol. Model. 2011, 17, 2305.doi: 10.1007/s00894-011-1059-7
(9) Zhou, C.; Yao, S.; Wu, J.; Chen, L.; Forrey, R. R.; Cheng, H. J.Comput. Theor. Nanosci. 2009, 6, 1. doi: 10.1166/jctn.2009.1181
(10) Szarek, P.; Urakami, K.; Zhou, C.; Cheng, H.; Tachibana, A.J. Chem. Phys. 2009, 130, 084111. doi: 10.1063/1.3072369
(11) Chen, L.; Zhou, C. G.; Wu, J. P.; Cheng, H. S. Front. Phys. China 2009, 4, 356. doi: 10.1007/s11467-009-0050-6
(12) Zhou, C.; Yao, S.; Wu, J.; Forrey, R. C.; Chen, L.; Tachibana, A.;Cheng, H. Phys. Chem. Chem. Phys. 2008, 10, 5445.doi: 10.1039/B804877K
(13) Zhou, C.; Wu, J.; Nie, A.; Forrey, R. C.; Tachibana, A.; Cheng, H.J. Phys. Chem. C 2007, 111, 12773. doi: 10.1021/jp073597e
(14) Godbey, D. J.; Somorjai, G. A. Surf. Sci. 1988, 204, 301.doi: 10.1016/0039-6028(88)90215-4
(15) Christmann, K. Surf. Sci. Rep. 1988, 9, 1.doi: 10.1016/0167-5729(88)90009-X
(16) Papoian, G.; Nørskov, J. K.; Hoffmann, R. J. Am. Chem. Soc. 2000,122, 4129. doi: 10.1021/ja993483j
(17) Hammer, B.; Norskov, J. K. Nature 1995, 376, 238.doi: 10.1038/376238a0
(18) Olsen, R. A.; Kroes, G. J.; Baerends, E. J. J. Chem. Phys. 1999, 111,11155. doi: 10.1063/1.480473
(19) Watson, G. W.; Wells, R. P. K.; Willock, D. J.; Hutchings, G. J.J. Phys. Chem. B 2001, 105, 4889. doi: 10.1021/jp002864c
(20) Nobuhara, K.; Kasai, H.; Diño, W. A.; Nakanishi, H. Surf. Sci. 2004,566–568, Part 2, 703. doi: 10.1016/j.susc.2004.06.003
(21) Nobuhara, K.; Kasai, H.; Nakanishi, H.; Okiji, A. J. Appl. Phys.2002, 92, 5704. doi: 10.1063/1.1512965
(22) Deng, J.; Li, H.; Xiao, J.; Tu, Y.; Deng, D.; Yang, H.; Tian, H.; Li, J.;Ren, P.; Bao, X. Energy Environ. Sci. 2015, 8, 1594.doi: 10.1039/c5ee00751h
(23) Wei, H.; Liu, X.; Wang, A.; Zhang, L.; Qiao, B.; Yang, X.; Huang,Y.; Miao, S.; Liu, J.; Zhang, T. Nat. Comm. 2014, 5, 5634.doi: 10.1038/ncomms6634
(24) Shin, S. I.; Go, A.; Kim, I. Y.; Lee, J.; Lee, Y.; Hwang, S.-J. Energy Environ. Sci. 2013, 6, 608. doi: 10.1039/c2ee22739h
(25) Lei, Y.; Mehmood, F.; Lee, S.; Greeley, J.; Lee, B.; Seifert, S.;Winans, R. E.; Elam, J. W.; Meyer, R. J.; Redfern, P. C.; Teschner,D.; Schlogl, R.; Pellin, M. J.; Curtiss, L. A.; Vajda, S. Science 2010,328, 224. doi: 10.1126/science.1185200
(26) Corma, A.; Serna, P.; Concepcion, P.; Juan Calvino, J. J. Am. Chem.Soc. 2008, 130, 8748. doi: 10.1021/ja800959g
(27) Liu, X. Y.; Wang, A.; Zhang, T.; Mou, C.-Y. Nano Today 2013, 8,403. doi: 10.1016/j.nantod.2013.07.005
(28) Campbell, C. T. Nat. Chem. 2012, 4, 597. doi: 10.1038/nchem.1412
(29) Vayssilov, G. N.; Lykhach, Y.; Migani, A.; Staudt, T.; Petrova, G. P.;Tsud, N.; Skala, T.; Bruix, A.; Illas, F.; Prince, K. C.; Matolin, V.;Neyman, K. M.; Libuda, J. Nat. Mater. 2011, 10, 310.doi: 10.1038/nmat2976
(30) Anderson, P. E.; Rodriguez, N. M. Chem. Mater. 2000, 12, 823.doi: 10.1021/cm990582n
(31) Barrio, L.; Liu, P.; Rodríguez, J. A.; Campos-Martín, J. M.; Fierro, J.L. G. J. Chem. Phys. 2006, 125, 164715. doi: 10.1063/1.2363971
(32) Liu, Z.-P.; Hu, P. J. Am. Chem. Soc. 2003, 125, 1958.doi: 10.1021/ja0207551
(33) Gong, X.-Q.; Selloni, A.; Dulub, O.; Jacobson, P.; Diebold, U. J. Am.Chem. Soc. 2008, 130, 370. doi: 10.1021/ja0773148
(34) Liu, X.; Dilger, H.; Eichel, R. A.; Kunstmann, J.; Roduner, E.J. Phys. Chem. B 2006, 110, 2013. doi: 10.1021/jp0561874
(35) Okamoto, Y. Chem. Phys. Lett. 2005, 405, 79.doi: 10.1016/j.cplett.2005.02.018
(36) Okamoto, Y. Chem. Phys. Lett. 2006, 429, 209.doi: 10.1016/j.cplett.2006.08.013
(37) Gdowski, G. E.; Fair, J. A.; Madix, R. J. Surf. Sci. 1983, 127, 541.doi: 10.1016/0039-6028(83)90046-8
(38) Richter, L. J.; Ho, W. Phys. Rev. B 1987, 36, 9797.doi: 10.1103/PhysRevB.36.9797
(39) Au, C. T.; Zhou, T. J.; Lai, W. J. Catal. Lett. 1999, 62, 147.doi: 10.1023/a:1019019710780
(40) Watari, N.; Ohnishi, S. J. Chem. Phys. 1997, 106, 7531.doi: 10.1063/1.473751
(41) Koszinowski, K.; Schroder, D.; Schwarz, H. J. Phys. Chem. A 2003,107, 4999. doi: 10.1021/jp027713j
(42) Swart, I.; Fielicke, A.; Redlich, B.; Meijer, G.; Weckhuysen, B. M.;De Groot, F. M. F. J. Am. Chem. Soc. 2007, 129, 2516.doi: 10.1021/ja066261b
(43) Swart, I.; De Groot, F. M. F.; Weckhuysen, B. M.; Gruene, P.; Meijer,G.; Fielicke, A. J. Phys. Chem. A 2008, 112, 1139.doi: 10.1021/jp076702t
(44) Wang, L. S.; Cheng, H. S.; Fan, J. W. J. Chem. Phys. 1995, 102,9480. doi: 10.1063/1.468817
(45) Hakkinen, H.; Yoon, B.; Landman, U.; Li, X.; Zhai, H. J.; Wang, L.S. J. Phys. Chem. A 2003, 107, 6168. doi: 10.1021/jp035437i
(46) Castleman, A. W.; Keesee, R. G. Chem. Rev. 1986, 86, 589.doi: 10.1021/cr00073a005
(47) Deheer, W. A. Rev. Mod. Phys. 1993, 65, 611.doi: 10.1103/RevModPhys.65.611
(48) Kerpal, C.; Harding, D. J.; Rayner, D. M.; Fielicke, A. J. Phys.Chem. A 2013, 117, 8230. doi: 10.1021/jp405120u
(49) Szarek, P.; Urakami, K.; Zhou, C.; Cheng, H.; Tachibana, A.J. Chem. Phys. 2009, 130, doi: 10.1063/1.3072369
(50) Chen, L.; Cooper, A. C.; Pez, G. P.; Cheng, H. J. Phys. Chem. C 2007, 111, 5514. doi: 10.1021/jp070181s
(51) Nie, A.; Wu, J.; Zhou, C.; Yao, S.; Luo, C.; Forrey, R. C.; Cheng, H.Int. J. Quantum Chem. 2007, 107, 219. doi: 10.1002/qua.21011
(52) Luo, C.; Zhou, C.; Wu, J.; Kumar, T. J. D.; Balakrishnan, N.; Forrey,R. C.; Cheng, H. Int. J. Quantum Chem. 2007, 107, 1632.doi: 10.1002/qua.21315
(53) Barreteau, C.; Guirado-López, R.; Spanjaard, D.; Desjonquères, M.C.; Oleś, A. M. Phys. Rev. B 2000, 61, 7781.doi: 10.1103/PhysRevB.61.7781
(54) Li, J. N.; Pu, M.; Ma, C. C.; Tian, Y.; He, J.; Evans, D. G. J. Mol.Catal. A: Chem. 2012, 359, 14. doi: 10.1016/j.molcata.2012.03.015
(55) Kadioglu, Y.; Demirkiran, A.; Yaraneri, H.; Akturk, O. U. J. Alloy.Compd. 2014, 591, 188. doi: 10.1016/j.jallcom.2013.12.074
(56) Ignatov, S. K.; Okhapkin, A. I.; Gadzhiev, O. B.; Razuvaev, A. G.;Kunz, S.; Baumer, M. J. Phys. Chem. C 2016, 120, 18570.doi: 10.1021/acs.jpcc.6b04555
(57) Liu, X. J.; Tian, D. X.; Meng, C. G. J. Mol. Struct. 2015, 1080, 105.doi: 10.1016/j.molstruc.2014.09.078
(58) Pelzer, A. W.; Jellinek, J.; Jackson, K. A. J. Phys. Chem. A 2015,119, 3594. doi: 10.1021/jp512643a
(59) Shi, Y.; Ervin, K. M. J. Chem. Phys. 1998, 108, 1757.doi: 10.1063/1.475608
(60) Balteanu, I.; Balaj, O. P.; Beyer, M. K.; Bondybey, V. E. Phys. Chem.Chem. Phys. 2004, 6, 2910. doi: 10.1039/b405211k
(61) Huang, L.; Han, B.; Xi, Y. J.; Forrey, R. C.; Cheng, H. S. ACS Catal.2015, 5, 4592. doi: 10.1021/acscatal.5b00689
(62) Harding, D. J.; Kerpal, C.; Meijer, G.; Fielicke, A. Angew. Chem. Int.Ed. 2012, 51, 817. doi: 10.1002/anie.201107042
(63) Helali, Z.; Markovits, A.; Minot, C.; Abderrabba, M. Chem. Phys.Lett. 2013, 565, 45. doi: 10.1016/j.cplett.2013.02.026
(64) Bruix, A.; Rodriguez, J. A.; Ramirez, P. J.; Senanayake, S. D.; Evans,J.; Park, J. B.; Stacchiola, D.; Liu, P.; Hrbek, J.; Illas, F. J. Am.Chem. Soc. 2012, 134, 8968. doi: 10.1021/ja302070k
(65) Kerpal, C.; Harding, D. J.; Rayner, D. M.; Fielicke, A. J. Phys.Chem. A 2013, 117, 8230. doi: 10.1021/jp405120u
Nickel Family Metal Clusters for Catalytic Hydrogenation Processes
HAN Bo CHENG Han-Song*
(Sustainable Energy Laboratory, Faculty of Materials Science and Chemistry, China University of Geosciecnes (Wuhan),Wuhan 430074, P. R. China)
Nanoparticles of precious metals play an important role in many heterogeneous catalytic reactions due to their excellent catalytic performance. As an idealized model, gas phase metal clusters have been extensively utilized to understand catalytic mechanisms at a molecular level. Here we provide an overview of our recent studies on H2dissociative chemisorption on nickel family clusters. The structure evolution and the stability of the metal clusters were first compared. H2dissociation on the clusters was then carefully addressed to understand the capability of metal clusters to break the H-H bond. Two key parameters, the dissociative chemisorption energy (ΔECE) and the H sequential desorption energy (ΔEDE),were employed to characterize the catalytic activity of metal clusters. Our results show that both ΔECEand ΔEDEdecline significantly as the H coverage increases. Since the catalyst is in general covered entirely by H atoms and H2molecules in a typical hydrogenation process, and maintained at a pre-determined pressure of H2gas, we can rationally use the calculated ΔECEand ΔEDEvalues at full H saturation to address the activity of metal clusters. Our results suggest that at full H coverage, each Pt atom is essentially capable of adsorbing 4 H atoms, while each Ni or Pd atom can only accommodate 2 H atoms.Considering the similar values of H desorption energies on Pt and Pd clusters, the higher average H capacity per Pt atom could probably lead to a faster reaction rate because more active H atoms are produced on the Pt catalyst particles in the hydrogenation process. Finally, the charge sensitivity of the key catalytic properties of Pt clusters for hydrogenation was systematically evaluated. The results show that the dissociation of H2and H desorption are strongly correlated to the charge state of the Pt clusters at low H coverage. However, at high H-capacities, both ΔECEand ΔEDEfall into a narrow range, suggesting that the charge can be readily dispersed and that the Pt―H bonds average the interaction between clusters and H atoms. As a result, the H-capacities on charged clusters were found to be similar as the cluster size increased; in case of sufficiently large clusters, the reactivity of a fully saturated cluster was no longer sensitive to its charge state.
Clusters; Transition metal; Catalytic hydrogenation; Full H saturation; Charge state; Density functional theory
December 12, 2016; Revised: April 5, 2017; Published online: April 17, 2017.
O641
10.1021/ic062183h
[Feature Article]
10.3866/PKU.WHXB201704172 www.whxb.pku.edu.cn
*Corresponding author. Email: chghs2@gmail.com; Tel: +86-27-67884283.
The project was supported by the National Natural Science Foundation of China (21473164, 21203169, 21233006) and the Fundamental Research Funds for the Central Universities, China University of Geosciences, China, and Air Products and Chemicals, Inc.
国家自然科学基金(21473164, 21203169, 21233006), 中国地质大学(武汉)中央高校基本科研业务费以及空气与化学品公司资助项目
© Editorial office of Acta Physico-Chimica Sinica
HAN Bo is an Associate Professor of Chemistry at China University of Geosciences Wuhan. His current research interests focus on computational chemistry and materials, including surface chemistry,energy materials, homogeneous and heterogeneous catalysis.
CHENG Han-Song is a Professor of Chemistry at China University of Geosciences Wuhan. In 2009, he was inducted into the “National 1000 Talents Plan” program in China and has served as the director of Sustainable Energy Laboratory at the university since then.His research interest lies in the area of first principles simulations and experimental development of novel materials for gas storage and separation, catalysis, battery electrolytes,and proton exchange membrane fuel cells. He is an author of over 200 peer-reviewed publications and an inventor of over 50 U.S. patents and patent applications.
猜你喜欢
科学技术创新(2022年30期)2022-10-21
农业与技术(2021年23期)2021-12-14
农业资源与环境学报(2021年4期)2021-07-30
黑龙江水利科技(2020年8期)2021-01-21
云南化工(2020年11期)2021-01-14
装备维修技术(2020年5期)2020-11-20
矿产综合利用(2020年1期)2020-07-24
少儿美术(快乐历史地理)(2020年2期)2020-06-22
安全(2020年3期)2020-04-25
原子与分子物理学报(2020年5期)2020-03-17
