
药品审评制度改革在路上
2017-10-29 10:58国家食品药品监督管理总局药品审评中心
中国卫生 2017年5期
2016年,国家食品药品监督管理总局药品审评中心(以下简称药审中心)围绕《国务院关于改革药品医疗器械审评审批制度的意见》(国发〔2015〕44号)相关要求,不断推进审评制度改革,全年完成审评并呈送总局审批的注册申请共12068件(以受理号计,下同),完成数量较2015年提高了26%,基本消除了注册积压。化药和疫苗临床试验申请、中药民族药各类注册申请已基本实现按时限审评,达成了阶段性目标。
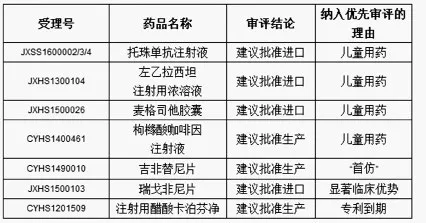
已完成审评建议批准上市的优先审评注册申请名单
新药和重要领域药物获优先审评审批
根据《关于解决药品注册申请积压实行优先审评审批的意见》(食药监药化管〔2016〕19号,以下简称19号文),药审中心共将12批193件注册申请纳入优先审评程序(中药注册申请两件、化药注册申请169件、生物制品注册申请22件)。其中,具有明显临床价值的新药注册申请共85件,占44%。截至2016年年底,纳入优先审评程序的注册申请中已有57件完成审评,其中,有11件为建议批准上市(含原料药注册申请两件)。
2016年,国家食品药品监督管理总局批准了206件药品生产(上市)注册申请(中药两件、化学药品188件、生物制品16件),批准了3666件药物临床试验注册申请(中药84件、化学药品3311件、生物制品271件)。 一批具有明显临床价值的创新药、临床急需药、专利到期药和我国首仿药也完成审评并建议批准上市。包括抗肿瘤药物瑞戈非尼片、培唑帕尼片、吉非替尼片;抗感染药物苹果酸奈诺沙星胶囊、富马酸贝达喹啉片、富马酸替诺福韦二吡呋酯片、聚乙二醇干扰素α2b注射液;风湿性疾病及免疫药物托珠单抗注射液;内分泌系统药物贝那鲁肽注射液;呼吸系统疾病及抗过敏药物金花清感颗粒预防用生物制品(疫苗)13价肺炎球菌结合疫苗。这些重要领域治疗药物的获批为患者提供了新的治疗手段,吉非替尼片、富马酸替诺福韦二吡呋酯片等国产首个仿制药也同时保障了患者用药可及性与可支付性。
审评发现不少问题
在2016年的药品审评过程中,有2139件注册申请的审评结论为建议不批准,1654件注册申请的第1轮审评结论为补充资料。这些未通过审评的情况集中反映出各类药品注册申请在研究和申报过程中存在的主要问题。
创新药新药临床试验(IND)前期的安全性研究不够充分或研究数据可靠性不足,临床方案中对受试者风险管控措施不足或整体设计欠完善;新药上市申请(NDA)临床试验规范性差,数据质量较差,临床试验结果可靠性不足,NDA申报资料中生产工艺信息不够详细的问题也较为常见;仿制药药学工艺、质量标准等研究存在较大缺陷,稳定性研究存在不足;前期研究不够充分,与审评要求差距过大,导致申请人未能按期完成补充资料或在后期主动放弃补充资料;进口上市注册的申报资料未提供国外上市的全部研究数据,关键信息缺失,资料翻译错误较多,可读性差;进口再注册申请未按批件要求完成上市后研究。

2018年实现按规定时限审评
2016年,药审工作取得了一定进展,但仍存在一些问题:一是造成注册审评积压的体制性、机制性问题还未从根本上解决;二是审评能力与医药产业创新发展、转型升级的需求还不适应;三是审评管理制度和标准体系仍需进一步完善;四是审评基础工作仍较薄弱,历史遗留问题尚未完全解决。
针对这些问题,药审中心今年将进一步加大解决药品审评积压力度,确保本年度完成注册申请积压,2018年实现按规定时限审评;围绕提高审评质量,鼓励创新,增加审评透明度,继续深化、细化、实化优先审评、沟通交流、项目管理、适应症团队、专家咨询、信息公开、立卷审查制度等工作;加快建立和完善审评质量控制体系、技术指南体系、合规管理体系;加快推进药品生产工艺登记核对工作,建立工艺登记平台,出台工艺变更指导原则,规范工艺变更管理;建立药品品种档案,建立完善包括药品工艺、处方、原辅料包材、质量标准、说明书、上市后安全信息、生产工艺变化等信息的数据库;建立药品电子通用技术文档(eCTD)系统,争取本年底前实现化学仿制药按eCTD要求接收申报和进行审评;进一步优化内设组织机构,提升科学化管理水平;严格审评人员的管理,加强保密制度建设和监督管理;优化审评员职业发展体系。
猜你喜欢
猪业科学(2022年10期)2022-11-03
吉林畜牧兽医(2022年7期)2022-07-20
医学食疗与健康(2022年2期)2022-04-23
猪业科学(2022年2期)2022-04-21
猪业科学(2022年1期)2022-03-24
皮肤病与性病(2021年3期)2021-07-30
皮肤病与性病(2021年3期)2021-07-30
中华养生保健(2020年5期)2020-11-16
当代水产(2020年4期)2020-06-16
环球时报(2019-06-26)2019-06-26
