
p38 MAPK通路参与舒巴坦诱导的大鼠脑缺血耐受
2017-10-19 03:09羡晓辉高俊侠李文斌
河北医科大学学报 2017年10期
羡晓辉,高俊侠,齐 杰,李文斌
(河北医科大学基础医学院病理生理学教研室,河北 石家庄 050017)
·论著·
p38 MAPK通路参与舒巴坦诱导的大鼠脑缺血耐受
羡晓辉,高俊侠,齐 杰,李文斌*
(河北医科大学基础医学院病理生理学教研室,河北 石家庄 050017)
目的探讨p38 MAPK信号通路在舒巴坦诱导大鼠脑缺血耐受中的作用。方法25只无特定病原体的Wistar大鼠分为5组,每组5只。①假手术组:侧脑室注射生理盐水10 μL,随后行全脑缺血的假手术,手术只暴露和分离双侧椎动脉及双侧颈总动脉,不阻断其血流;②全脑缺血组:首先侧脑室注射生理盐水10 μL,随后即刻行8 min全脑缺血手术;③舒巴坦+全脑缺血组:首先给大鼠侧脑室注射舒巴坦溶液(360 nmol,10 μL),随后即刻行8 min全脑缺血手术;④SB203580+舒巴坦+全脑缺血组:首先给大鼠侧脑室注射SB203580溶液(5 nmol,10 μL),30 min后给予舒巴坦(360 nmol,10 μL)侧脑室注射,随后即刻行8 min全脑缺血手术;⑤SB203580组:首先给大鼠侧脑室注射SB203580(5 nmol,10 μL),30 min后按照假手术组的方法,注射生理盐水及行脑缺血假手术。末次手术后7 d处死动物,取脑组织进行硫堇染色,观察大鼠海马CA1区神经细胞的形态和神经细胞密度,并进行组织分级。结果全脑缺血组大鼠海马CA1区大片神经细胞破坏、死亡,细胞排列疏松、紊乱,散在大量细胞碎片,神经细胞密度降低明显,组织学分级明显升高;舒巴坦+全脑缺血组组织学分级明显减低,神经细胞密度明显升高; SB203580+舒巴坦+全脑缺血组神经细胞密度明显降低,组织学分级明显升高。结论p38 MAPK参与了舒巴坦预处理诱导的大鼠脑缺血耐受。
缺氧,脑;舒巴坦;大鼠
随着中国社会老龄化程度日益加重以及社会生活、工作节奏的日益加快,缺血性脑损伤在临床呈现多发势态,严重威胁着大众的健康。缺血性脑损伤的机制复杂[1],其中兴奋性氨基酸的大量释放造成的兴奋性神经毒性作用在缺血性脑损伤发病过程中占有重要地位[2]。突触间隙内谷氨酸的清除主要依赖高亲和性的兴奋性氨基酸转运系统进行,尤其是胶质细胞上的谷氨酸转运体(glialglutamatetransporter-1,GLT-1),在很多脑区的谷氨酸稳态维持中发挥重要作用。因此,调制脑内GLT-1的功能是缺血性脑损伤防治的重要策略之一。研究表明,β内酰胺类抗生素如头孢曲松钠可以上调大鼠海马CA1区GLT-1的表达,从而使神经细胞提高对脑缺血的耐受性[3],但由于头孢曲松钠为临床一线抗生素,大量应用会出现诸如菌群失调、耐药菌株出现、严重胃肠道反应等,限制了其在临床缺血性脑损伤防治中的应用。舒巴坦也属于β内酰胺类药物,且抗菌作用弱,不良反应小。本研究应用大鼠全脑缺血模型,观察舒巴坦预处理对全脑缺血再灌注引起的海马CA1区神经细胞迟发性死亡的影响,并进一步应用SB203580抑制p38MAPK通路活性,以探讨p38MAPK通路在舒巴坦诱导的大鼠脑缺血耐受中的作用。
1 材 料 与 方 法
1.1 实验动物及试剂 健康雄性无特定病原体Wistar大鼠25只,体质量260~300g,购自河北省实验动物中心。所有实验动物单笼饲养,自由食水,在实验室适应环境2d后用于实验。动物实验操作流程符合河北医科大学动物实验操作规范。主要试剂:舒巴坦标准品(中国食品药品检定研究院),硫堇(美国Sigma),SB203580 (美国PeproTech)。
1.2 实验分组 各组动物分别侧脑室预置给药套管后,随机分为以下5组,每组5只。①假手术(sham)组:首先侧脑室注射生理盐水 10μL,随后即刻行全脑缺血的假手术;②全脑缺血组(ischemia):首先侧脑室注射生理盐水 10μL,随后即刻行8min全脑缺血手术;③舒巴坦+全脑缺血组(sulbactam+ischemia):首先给大鼠侧脑室注射舒巴坦溶液(360nmol,10μL),随后即刻行8min全脑缺血手术。④SB203580+舒巴坦+全脑缺血组(SB203580+sulbactam+ischemia):首先给大鼠侧脑室注射SB203580溶液(5nmol,10μL),30min后给予舒巴坦(360nmol,10μL)侧脑室注射,随后即刻行8min全脑缺血手术,方法同舒巴坦+脑缺血组;⑤SB203580+假手术组(SB203580+sham):首先给大鼠侧脑室注射SB203580(5nmol,10μL),30min后按照假手术组方法,注射生理盐水及行脑缺血假手术。各组动物于全脑缺血假手术或全脑缺血再灌注后7d断头处死,进行脑组织病理学观察,评价神经细胞的存活情况。
1.3 手术操作及模型建立
1.3.1 侧脑室置管与注射 实验中舒巴坦、SB203580均采用侧脑室注射给药。首先进行大鼠侧脑室置管,小动物麻醉机给予异氟烷持续吸入,在脑立体定位仪上进行固定,选用俯卧位,于大鼠颅骨顶部正中切开皮肤,刮剥颅顶骨膜,双氧水局部处理,保持颅骨骨面干燥,参照大鼠脑解剖图谱,确定侧脑室定位位置,于前囟向右侧1.5mm、向后0.8mm,在颅骨上用牙科钻开孔,将给药套管经孔置入,套管插管深度3.8~4.0mm,牙科水泥固定,作为侧脑室给药的入路。术后动物单笼饲养,自由食水。动物恢复7d后,待手术应激消除、血脑屏障修复后,用于进一步实验。
按照实验时间点,使用PE连接管连接微量注射器和套管注射内芯,旋开套管螺帽并拔出,将注射内芯插入到侧脑室套管内,利用微量注射泵推动针栓给药,10min完成注射,留针10min,防止药物溢出。注射过程中注意安抚动物,使之保持安静。注射结束后旋紧套管螺帽,继续进行下一步实验。
1.3.2 全脑缺血模型的制备 参考Pulsinelli等[4]的方法,应用小动物麻醉机给予大鼠异氟烷持续吸入,取俯卧位固定动物,于枕骨后背侧,沿颈部正中1.5cm纵形切口,钝性分离颈旁肌,充分暴露出第一颈椎翼状孔,用预热的电灼针在翼状孔中烧灼,凝闭双侧椎动脉。用庆大霉素注射液冲洗切口,间断缝合关闭切口。术后待动物恢复2d后,选用恢复良好的动物进行全脑缺血的下一步实验。
椎动脉凝闭的大鼠在异氟烷麻醉下,仰卧位固定,颈部正中纵行开口,分离双侧颈总动脉。停止异氟烷吸入,待动物清醒后用动脉夹阻断双侧颈总动脉血流,行成全脑缺血,持续8min后恢复双侧颈总动脉血流灌注。庆大霉素冲洗,缝合切口,回笼单笼饲养,于规定时间点进行处死、取材。手术过程中,白炽灯照射以维持动物体温。成功形成全脑缺血的标准为:大鼠双侧颈总动脉被夹闭后,出现意识丧失,双侧瞳孔对光反射消失,瞳孔散大80%~100%,动物翻正反射消失。在缺血期中,上述意识丧失或神经反射的改变均未恢复,动物保持平稳呼吸,无抽搐,术后无偏瘫或癫痫。全脑缺血的Sham手术只暴露和分离双侧椎动脉及双侧颈总动脉,不阻断其血流,其余操作同全脑缺血手术。
1.4 脑组织病理学评价 快速剥离大脑,于视交叉后1~3mm冠状切取脑组织,4%多聚甲醛固定,石蜡包埋,5μm组织切片,梯度脱蜡置水后进行组织切片硫堇染色,比较大鼠海马CA1区神经细胞的神经细胞密度(neuronaldensity,ND)和组织学分级(histologicalgrading,HG)改变,以确定不同条件下神经细胞迟发性死亡情况。
1.4.1HG参照Kitagawa等[5]和Kato等[6]建立的神经系统组织学分级方法,光学显微镜下观察,对海马CA1区神经细胞的病理学改变确定分级。分级标准如下:0级,目标区域未见神经细胞死亡;Ⅰ级,目标区域可见散在神经细胞死亡;Ⅱ级,目标区域可见大片神经细胞死亡;Ⅲ级,目标区域几乎全部神经细胞死亡。每只动物选择3张切片,每张组织切片双侧均进行分级,采用双侧的平均等级为最终HG结果进行统计。
1.4.2ND在光学显微镜(40×)下观察海马CA1区,对每1mm2区段内细胞膜完整、细胞核饱满且核仁清晰的神经细胞数目进行计数,每只动物选择3张切片,每张组织切片两侧海马分别计数3个区段,取平均数作为ND值进行统计分析。
1.5 统计学方法 应用Prism6.0统计软件分析数据。计量资料比较分别采用F检验和q检验。P<0.05为差异有统计学意义。
2 结 果
2.1 各组大鼠海马CA1区神经细胞的形态学改变 假手术组:海马CA1区锥体神经细胞约为3~4层,整齐、致密排布,可见清晰、完整的细胞轮廓,胞内有大量尼氏体着色,细胞核充实饱满,核仁清晰(图1A)。全脑缺血组:8min全脑缺血引起了海马CA1区明显的神经细胞损伤,镜下可见大片神经细胞破坏、死亡,细胞排列疏松、紊乱,散在大量细胞碎片,残存的细胞收缩,呈不规则状,核浓染,核仁消失(图1B)。舒巴坦+全脑缺血组:仅有散在细胞死亡,大部分细胞可见清晰、完整的细胞轮廓,整齐排列,层次清楚(图1C)。SB203580+舒巴坦+缺血组:阻断了舒巴坦介导的神经保护作用,镜下可见海马CA1区分布着大量死亡的神经细胞碎片及不规则形状的残存细胞,细胞核深染,核仁消失(图1D)。SB203580 + 假手术组:未造成大鼠海马CA1区的神经细胞的明显改变(图1E)。
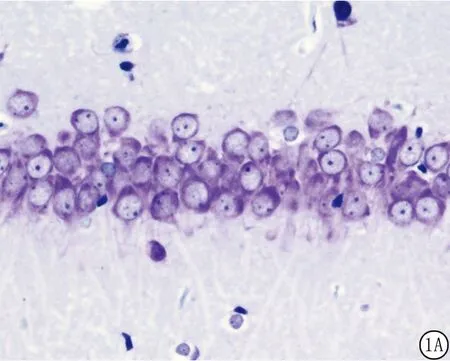
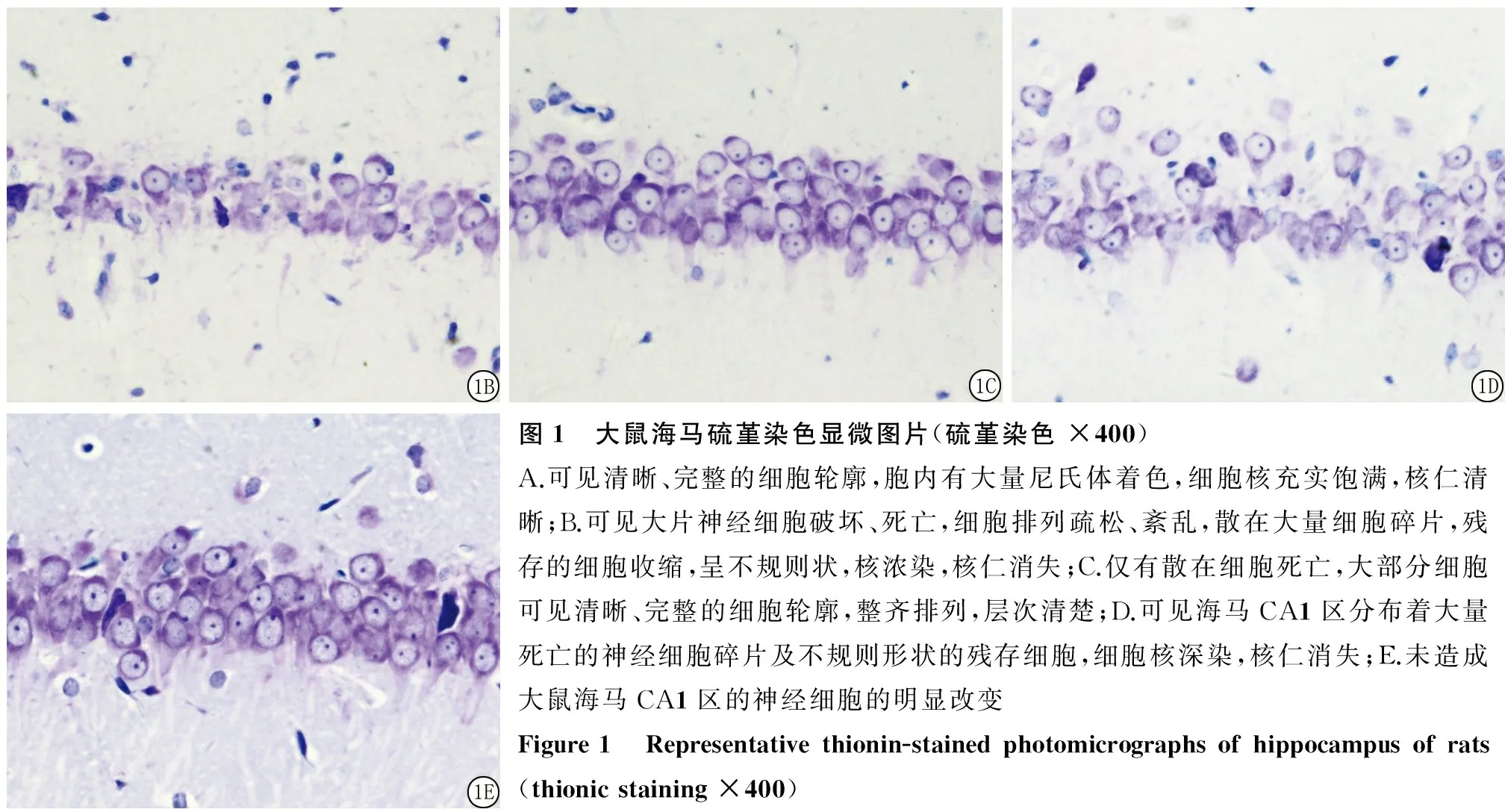
图1 大鼠海马硫堇染色显微图片(硫堇染色×400)A.可见清晰、完整的细胞轮廓,胞内有大量尼氏体着色,细胞核充实饱满,核仁清晰;B.可见大片神经细胞破坏、死亡,细胞排列疏松、紊乱,散在大量细胞碎片,残存的细胞收缩,呈不规则状,核浓染,核仁消失;C.仅有散在细胞死亡,大部分细胞可见清晰、完整的细胞轮廓,整齐排列,层次清楚;D.可见海马CA1区分布着大量死亡的神经细胞碎片及不规则形状的残存细胞,细胞核深染,核仁消失;E.未造成大鼠海马CA1区的神经细胞的明显改变Figure1 Representativethionin-stainedphotomicrographsofhippocampusofrats(thionicstaining×400)
2.2 各组大鼠海马CA1区神经细胞组织学分级比较 全脑缺血组大鼠海马CA1区神经细胞组织学分级较假手术组明显升高;与全脑缺血组比较,舒巴坦+全脑缺血组组织学分级明显降低;与舒巴坦+全脑缺血组相比较,SB203580+舒巴坦+全脑缺血组应用SB203580后,组织学分级明显升高;SB203580+假手术组组织学分级与假手术组无明显差别。见图2。
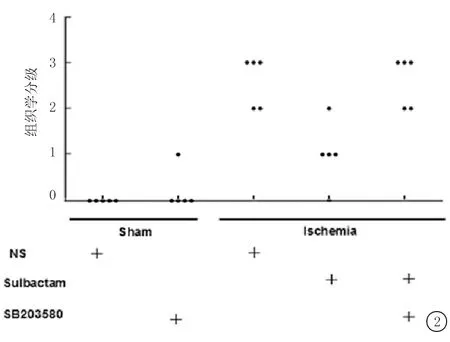
图2大鼠海马CA1区组织学分级
Figure2ThehistologicalgradeintheCA1hippocampusofrats
2.3 各组大鼠海马CA1区神经细胞密度比较 全脑缺血组大鼠海马CA1区ND值较假手术组明显降低(P<0.05);与全脑缺血组比较,舒巴坦+全脑缺血组ND值明显升高(P<0.05);与舒巴坦+全脑缺血组相比较,SB203580+舒巴坦+全脑缺血组ND值明显降低(P<0.05)。 SB203580+假手术组海马CA1区ND值未见明显变化,与假手术组比较差异无统计学意义(P>0.05)。见表1。
表1 表1 各组大鼠海马CA1区神经细胞密度比较Table 1 Comparison of neuronal density in the CA1 hippocampus of rats among groups
*P<0.05 与假手术组比较 #P<0.05 与全脑缺血组比较
△P<0.05与舒巴坦+全脑缺血组比较(q检验)
3 讨 论
脑缺血性损伤的发病机制复杂,涉及细胞能量代谢障碍、兴奋性氨基酸毒性作用、炎症反应过度[7]、细胞凋亡、细胞内外离子稳态破坏、NO失衡、自由基损伤[8]等多个方面。其中兴奋性氨基酸释放导致的兴奋性神经毒性作用被人们广泛关注[9]。脑血流的中断导致神经细胞能量代谢障碍[10],细胞膜功能下降,细胞内外离子稳态破坏(细胞内高Na+[11]、Ca2+[12],细胞外高K+),导致神经细胞去极化,突触前膜谷氨酸大量释放,同时细胞外高钾抑制了突触前膜对谷氨酸的再摄取[13-14],最终导致突触间隙内谷氨酸大量堆积,与突触后膜的相应受体结合,使神经细胞持续兴奋,导致神经细胞的损伤[15]。本研究结果显示,大鼠进行8 min全脑缺血后7 d,海马CA1区神经细胞大量死亡,细胞层次缺失,可见大量的细胞碎片,核浓染,固缩;与假手术组比较,全脑缺血组大鼠海马CA1区神经细胞组织学分级明显升高,ND明显降低。表明8 min的全脑缺血造成了大量神经细胞迟发性死亡。
在遭受缺血打击时,主要分布于星形胶质细胞上的GLT-1的改变直接决定着神经细胞的存活与否,其功能的调制在脑缺血性疾病的发展中发挥着重要作用。脑缺血预处理可通过上调大鼠海马CA1区GLT-1的表达,提高突触间隙谷氨酸的清除能力,使神经细胞损伤减轻[16]。头孢曲松钠预处理[3]也可以上调GLT-1的表达而诱导脑缺血耐受,但是由于头孢曲松钠为临床一线抗生素,抗菌作用强大,临床大量使用会带来诸如菌群失调、耐药菌株出现、真菌感染、严重胃肠道反应等多种不良后果。这些不良反应限制了头孢曲松钠应用于防治临床缺血性脑损伤。舒巴坦也属于β内酰胺类药物,其化学结构中虽然也具有β内酰胺环,与头孢曲松钠类似,但其抗菌作用要远远弱于头孢曲松钠,且不良反应小,临床上一般与其他β内酰胺类抗生素联合用药,以增强抗生素的抗菌效应。在本研究中显示,预先给予舒巴坦预处理可以使神经细胞对随后发生的全脑缺血打击耐受性增强,大鼠海马CA1区神经细胞轮廓清晰,排列整齐,仅有少量细胞死亡;与全脑缺血组比较,其组织学分级明显降低,ND明显升高(P<0.05)。表明舒巴坦预处理也可以诱导大鼠脑缺血耐受,但其机制有待进一步深入研究。
p38 MAPK属于应激活化的蛋白激酶,是MAPKs家族的重要成员,细胞缺氧、活性氧、炎症因子增多等因素均可能激活p38 MAPK信号通路,参与多种病理生理过程。诸多研究显示p38 MAPK通路的大量活化参与了脑缺血性疾病时细胞的损伤过程。小鼠大脑中动脉阻塞后2 h缺血区的p38 MAPK活性明显增加,可呈现明显高峰[17]。大鼠全脑缺血时,在损伤严重的区域,p38 MAPK表达和活化明显增多[18],促进了炎症因子的增多,炎症因子又进一步激活p38 MAPK,两者相互协同,促进了缺血性脑病时神经细胞的死亡。而抑制p38 MAPK通路活性,可以使脑外伤大鼠的二次脑损伤发生概率大大降低[19]。通过预处理等方式调节p38 MAPK的表达,使其表达量适度增高也介导了缺血早期机体的内源性保护机制出现。Zhang等[20]报道,脑缺血预处理可以使大鼠海马CA1区p38 MAPK表达适度增高,介导GLT-1的表达增高,从而减轻随后的缺血打击造成的神经细胞迟发性死亡;抑制p38 MAPK的表达,这种神经保护作用也被阻断。上述研究结果表明p38 MAPK在脑缺血耐受的诱导过程中发挥着重要的作用。鉴于此,本研究中进一步探讨了p38 MAPK信号通路在舒巴坦诱导的脑缺血耐受中的作用,结果显示预先给予SB203580抑制p38 MAPK信号通路后,舒巴坦预处理诱导的这种神经保护作用也被抑制,海马CA1区分布着大量死亡的神经细胞碎片及不规则形状的残存细胞,细胞核深染,核仁消失;与舒巴坦+全脑缺血组相比,其组织学分级明显增高,ND明显减低。表明p38 MAPK信号通路可能参与了舒巴坦诱导的脑缺血耐受过程。
综上所述,p38 MAPK信号通路参与了舒巴坦预处理诱导大鼠脑缺血耐受,但由于p38 MAPK信号通路与脑缺血后的多种病理生理过程相关,故舒巴坦预处理时,p38 MAPK通路活化的下游机制有待进一步研究。
[1] An SJ,Kang TC,Park SK,et al. Oxidative DNA damage and alteration of glutamate transporter expressions in the hippocampal CA1 area immediately after ischemic insult[J]. Mol Cells,2002,13(3):476-480.
[2] Lipton P. Ischemic cell death in brain neurons[J]. Physiol Rev,1999,79(4):1431-1568.
[3] Hu YY,Xu J,Zhang M,et al. Ceftriaxone modulates uptake activity of glial glutamate transporter-1 against global brain ischemia in rats[J]. J Neurochem,2015,132(2):194-205.
[4] Pulsinelli WA,Buchan AM. The four-vessel occlusion rat model:method for complete occlusion of vertebral arteries and control of collateral circulation[J]. Stroke,1988,19(7):913-914.
[5] Kitagawa K,Matsumoto M,Tagaya M,et al. Ischemic tolerance'phenomenon found in the brain[J]. Brain Res,1990,528(1):21-24.
[6] Kato H,Liu Y,Araki T,et al. Temporal profile of the effects of pretreatment with brief cerebral ischemia on the neuronal damage following secondary ischemic insult in the gerbil:cumulative damage and protective effects[J]. Brain Res,1991,553(2):238-242.
[7] 温雅,董立鹏,赵景茹,等.旋覆花内酯通过抑制炎症反应对缺血脑组织发挥保护作用[J].河北医科大学学报,2016,37(5):497-500,505.
[8] 于沛霞,薄立军,薛辉,等.依达拉奉对脑缺血缺氧新生大鼠神经功能的影响[J].河北医科大学学报,2017,38(4):423-427.
[9] Robinson MB. The family of sodium-dependent glutamate transporters:a focus on the GLT-1/EAAT2 subtype[J]. Neurochem Int,1998,33(6):479-491.
[10] Rintoul GL,Filiano AJ,Brocard JB,et al. Glutamate decrease mitochondrial size and movement in primary forebrain neurons[J]. J Neurosci,2003,23(21):7881-7888.
[11] Matsumoto Y,Yamamoto S,Suzuki Y,et al. Na+/H+exchanger inhibitor,SM-20220,is protective against excitoxicity in cultured cortical neurons[J]. Stroke,2004,35(1):185-190.
[12] Nicotera P,Bano D. The enemy at the gates.Ca2+entry through TRPM7 channels and anoxic neuronal death[J]. Cell,2003,115(7):768-770.
[13] Whetsell WO Jr. Current concepts of excitotoxicity[J]. J Neuropathol Exp Neurol,1996,55(1):1-13.
[14] Bacigaluppi M,Russo GL,Peruzzotti-Jametti L,et al. Neural stem cell transplantation induces stroke recovery by upregulating glutamate transporter GLT-1 in astrocytes[J]. J Neurosci,2016,36(41):10529-10544.
[15] Gong HY,Zheng F,Zhang C,et al. Propofol protects hippocampal neurons from apoptosis in ischemic brain injury by increasing GLT-1 expression and inhibiting the activation of NMDAR via the JNK/Akt signaling pathway[J]. Int J Mol Med,2016,38(3):943-950.
[16] Zhang M,Li WB,Geng JX,et al. The upregulation of glial glutamate transporter-1 participates in the induction of brain ischemic tolerance in rats[J]. J Cereb Blood Flow Metab,2007,27(7):1352-1368.
[17] Wu DC,Ye W,Che XM,et al. Activation of mitogen-activated protein kinases after permanent cerebral artery occlusion in mouse brain[J]. J Cereb Blood Flow Metab,2000,20(9):1320-1330.
[18] Lennmyr F,Karlsson S,Gerwins P,et al. Activation of mitogen- activated protein kinases in experimental cerebral ischemia[J]. Acta Neurol Scand,2002,106(6):333-340.
[19] Yang H,Gu ZT,Li L,et al. SIRT1 plays a neuroprotective role in traumatic brain injury in rats via inhibiting the p38 MAPK pathway[J]. Acta Pharmacol Sin,2017,38(2):168-181.
[20] Zhang M,Gong JX,Wang JL,et al. p38 MAPK participates in the mediation of GLT-1 up-regulation during the induction of brain ischemic tolerance by cerebral ischemic preconditioning[J]. Mol Neurobiol,2017,54(1):58-71.
(本文编辑:许卓文)
p38MAPKsignalingpathwayparticipatesinthesulbactam-inducedbrainischemictoleranceinrats
XIANXiao-hui,GAOJun-xia,QIJie,LIWen-bin*
(DepartmentofPathophysiology,theCollegeofBasicMedicalScience,HebeiMedicalUniversity,Shijiazhuang050017,China)
ObjectiveTo investigate the role of p38 MAPK signaling pathway in sulbactam-induced brain ischemic tolerance in rats.MethodsThe model of rat global cerebral ischemia was used. After a stainless steel cannula was implanted in the right lateral ventricle,25 healthy male Wistar rats were randomly divided into the following groups(n=5 in each group). ①Sham group(normal saline+sham): the rats were administrated with normal saline of 10 μL via the cannula first, and then the sham operation for global cerebral ischemia was performed. ②Global cerebral ischemia group(ischemia): normal saline(10 μL) was administrated via the cannula first and then performed 8 minutes of global cerebral ischemia immediately. ③Sulbactam+ischemic group: sulbactam solution(10 μL, 360 nmol) was administrated via the cannula first and then performed 8 minutes of global cerebral ischemia immediately. ④SB203580+sulbactam+ischemic group: SB203580(10 μL, 5 nmol) was administrated via the cannula 30 minutes before sulbactam(10 μL, 360 nmol) and then performed 8 minutes of global cerebral ischemia immediately. ⑤SB203580+sham group: SB203580 solution(10 μL, 5 nmol) was administrated via the cannula first and then performed sham operation for global cerebral ischemia immediately. The neuropathological evaluation including histological grade(HG) and neuronal density(ND) was performed using the method of thionic staining to observe the survival situation of neurons in CA1 hippocampus.ResultsGlobal cerebral ischemia for 8 min induced obvious delayed neuronal death(DND)which resulted in large area of neuronal absence in the CA1 hippocampus with the significant increase in HG and decrease in ND. Pre-treatment with sulbactam effectively prevented the DND normally induced by the global brain ischemia. Compared with ischemic group,the HG was significantly reduced, and the ND increased significantly. Pretreatment with SB203580, an inhibitor of p38 MAPK signal pathway, blocked the neuroprotective effect mediated by sulbactam. Compared with sulbactam+ischemic group, the ND was significantly decreased, and the HG increased significantly.Conclusionp38 MAPK signaling pathway may participate in the sulbactam-induced brain ischemic tolerance in rats.
hypoxia,brain;sulbactam;rat
R845.22
A
1007-3205(2017)10-1117-06
2017-08-02;
2017-08-11
河北省高等学校科学技术研究青年基金项目(QN2015210)
羡晓辉(1978-),男,河北冀州人,河北医科大学基础医学院病理生理学教研室讲师,医学博士,从事神经病理生理学研究。
*通讯作者。E-mail:Liwbsjz@163.com
10.3969/j.issn.1007-3205.2017.10.001
猜你喜欢
中国药学药品知识仓库(2022年7期)2022-05-10
昆明医科大学学报(2021年6期)2021-07-31
医学食疗与健康(2021年27期)2021-05-13
中国兽医杂志(2019年2期)2019-06-25
中国妇幼健康研究(2019年2期)2019-03-26
中国组织化学与细胞化学杂志(2017年1期)2017-06-15
磁共振成像(2015年1期)2015-12-23
中国当代医药(2015年16期)2015-03-01
中国当代医药(2015年10期)2015-03-01
郑州大学学报(医学版)(2015年2期)2015-02-27
