
过渡金属层状双氢氧化物电催化剂研究进展
2017-10-13 06:37王志强刘卫红耿凤霞
中国材料进展 2017年9期
王志强,刘卫红,耿凤霞
(苏州大学材料与化学化工学部,江苏 苏州 215123)
过渡金属层状双氢氧化物电催化剂研究进展
王志强,刘卫红,耿凤霞
(苏州大学材料与化学化工学部,江苏 苏州 215123)
过渡金属层状双氢氧化物 (LDHs) 是一类典型的层状功能材料,由于结构、形貌和组成的广泛可调性以及独特的物理化学性质,使其在电化学能量储存和转化方面有着广泛的应用,尤其是在电催化水解领域发挥着重要的作用。目前对于设计同时具有低成本和高电化学活性与稳定性的过渡金属基电极材料仍然存在诸多的挑战,而LDHs材料的出现为设计这类材料提供了无限的可能。主要归纳了近期LDHs基电极材料在电催化水解中的研究进展。除此之外,也针对材料的设计和性能等方面存在的问题以及对未来的研究方向等进行了预测和展望。
层状双氢氧化物;二维材料;析氢反应;析氧反应;复合材料
Abstract:Transition-metal layered double hydroxides (LDHs), a class of layered functional material with high flexibility of structure, morphology and component, have been widely used in the field of electrochemical energy storage and conversion, especially in electrochemical water splitting. Currently, designing low-cost and earth-abundant transition-metal based electrode materials of high activity and electrochemical stability is urgently desired, for which LDHs may offer numerous opportunities. This review summarized the recent progresses on LDHs and their composites in the use of electrocatalyst for water splitting. In addition, current challenges and possible strategies were discussed based on the aspect of material design and performance.
Key words:layered double hydroxides; two-dimensional materials; hydrogen evolution reaction (HER); oxygen evolution reaction (OER); hybrid materials
1 前 言
随着对清洁可再生能源需求的增加,人类目前努力致力于发展高效的、低成本的和环境友好的新能源[1,2]。氢气是21世纪最清洁的能源,而电化学水解被认为是获得氢气最方便、最有效的方法之一。电解水通常包括两个半反应:析氢反应(HER)和析氧反应(OER)[3-5]。然而,要把电化学水解反应实际应用在工业上仍然具有一定的挑战性,最主要的原因在于要想获得一个理想的反应速率需要克服较高的反应能垒。因此为了降低过电势,加速反应获得有效的电催化剂是一个关键因素[6-8]。目前,贵金属氧化物RuO2和IrO2以及贵金属 Pt在OER和HER中具有较高的催化活性,然而由于他们极高的成本,限制了他们在工业中的应用[9-12]。因此发展低成本、资源丰富的过渡金属基材料已经受到了研究者们广泛的关注。由于过渡金属层状双氢氧化物的独特物理化学性质使其在析氢和析氧领域产生了巨大的潜力。在过去的数年里,已经报道了许多的过渡金属层状双氢氧化物基材料应用于电催化水解中[13-22]。
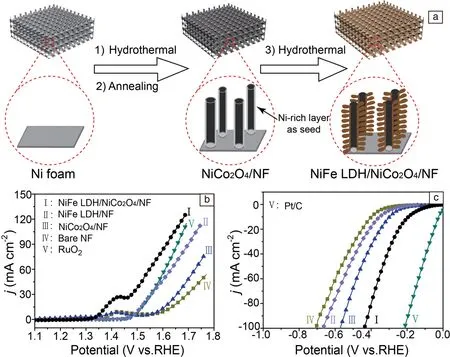
层状双金属氢氧化物也被称为类水滑石化合物,是层状功能材料中的重要一员。它的基本结构是由带正电的片层和与之电荷平衡的层间阴离子组成的三明治堆积结构。片层的结构类似于水镁石[Mg(OH)2], 其中片层内部的部分二价金属被三价金属取代,通用分子式为{(MII)1-x(MIII)x(OH)2}x+{(An-)x/n·yH2O}[23,24]。二价和三价金属的比例影响着层状双氢氧化物的电荷密度。x是三价阳离子的浓度,它对于LDHs的组成和最终的结构有着重要的影响。通常,当x值的范围在0.2 通常,LDHs的合成主要是通过对含两种不同价态的金属阳离子的盐溶液共沉淀得到。理论上任何与二价镁离子的半径接近的三价金属阳离子都能够形成LDHs主体层,但是对于过渡金属,尤其是三价铁离子,由于在较低的pH环境中容易形成溶胶状氢氧化物,导致通过共沉淀的方法很难形成过渡金属的LDHs。对此,可以通过加入少量的螯合剂进而均匀沉淀的方法成功解决了这个问题。另一方面,Ma等[29]通过尿素和六亚甲基四胺(HMT)在水溶液中加热,缓慢释放氢氧根,形成二价双金属的氢氧化物,随后通过拓扑化学氧化的方法,将M2+部分氧化M3+, 成功合成了具有高结晶性的含铁过渡金属LDHs, 这个方法的优点在于合理地控制氢氧化物中过渡金属的氧化态。此外,通过简单的电沉积的方法获得了各种组分LDHs基材料,如NiCo LDH, ZnCo LDH等[30]。 正是由于LDHs具有灵活的可调结构和丰富的合成方法,LDHs基材料可以应用于发展具有不同组成(片层主体和层间客体)的新型材料,设计不同的纳米结构(纳米片和多级纳米结构),构建不同的复合纳米材料等等, 进而使得LDHs在催化、燃料分解、电池、和电容器中有着广泛的应用[31, 32]。 本文将归纳近期基于不同成分和结构的LDHs基材料在电化学水解中的应用,包括剥离的纳米片、负载在导电基底上的超薄纳米片、与碳材料结合的复合材料、与功能化的无机材料结合的复合材料以及LDHs前体的转化(氧化、硫化、磷化和氨化)。希望未来会有更多的研究者关注LDH基材料在电化学水解中的应用并进一步去拓展这个领域。 由于贵金属较少的含量和在电催化中极高的应用成本,发展含量丰富的、具有较高活性和稳定性的过渡金属基催化剂变得越来越重要。过渡金属LDHs, 尤其是包含钴和镍的LDHs被认为是最理想的候选者[33-35]。而且他们表现出与贵金属相当的析氧反应速率。Song等[36]报道了超薄CoMn LDH纳米片在电催化析氧中的应用。在这个工作中,CoMn LDH是在室温下通过简单的共沉淀得到,同时具有非常低的过电势和较大的周转频率,是相应的Co和Mn氧化物、氢氧化物的20倍,是贵金属氧化铟的9倍。高的催化活性主要是由于在表面形成了无定型层以及在无定型层中四价活性钴的累积。随后,Song等[37]进一步报道了剥离的过渡金属LDH纳米片应用在电催化水解中。单片的LDH纳米片可以通过阴离子交换过程以及随后在甲酰胺中剥离获得。XRD测试证明了成功的阴离子交换和剥离过程。分析电化学测试的结果,表明所有成功剥离的LDH(NiFe, NiCo 和CoCo LDH)纳米片都要比相应的块体LDHs具有更高的催化活性。原因是这种剥离状态增加了比表面积、创造了更多的活性位点,同时提升了电导性。 2.2.1 LDH负载在导电基底上 对于单纯的LDHs导电性差,为此研究者将LDH直接生长在导电基底上,比如泡沫镍、钛片和碳布[38, 39]。常用的两种合成方法是电沉积和水热生长,Li等[30]通过简易电沉积的方法直接在导电基底上生长ZnCo LDH。合成的LDH纳米片垂直生长在泡沫镍上,形成相互交联的结构,从而产生优越的电解水氧化性能。在碱性环境中启动电势只有0.33 V, 周转频率也要高于单金属Co(OH)2。具有较高活性的主要原因是这种垂直生长的纳米片充分地暴露了活性位点,而且避免了使用粘接剂,提高了导电性。文献报道了[40]将NiFe LDH水热生长在泡沫镍上,实现了在碱性环境中双功能电解水,而且本文首次将太阳能电池与催化剂组装成一个器件,实现了太阳能向燃料的有效转化。太阳能电池可以在太阳光下产生较高的电压电流,将其与电催化材料连接在一起,在太阳光的照射下产生电压驱动,催化剂在电解质中发生反应从而产生氢能源。在图1b中将整个串联器件中电子的转移示意出来,通过这样一个串联的电路能够实现在10 mA/cm2的电流密度下达到太阳能电子功率转移率是12.3%,即使在高的电压2 V下也能实现太阳能电子功率转移率为15.7%。通过这篇文章的设计思路,有理由相信在未来实现不同器件的组装以及能源可持续发展的可靠性。 图1 水解器件的示意图 (a), 能级传输示意图 (b), 串联电池的J-V曲线 (c), 组装水解器件的电流密度-时间曲线 (d) [40]Fig.1 Schematic diagram of the water-splitting device (a),schematic of the device and the relevant energy levels (b), J-V curves of the perovskite tandem cell (c), Current density-time curve of the integrated water splitting device (d) [40] 2.2.2 LDH与碳材料的复合 如上所述,尽管之前已经成功合成了高结晶性的LDHs, 但是由于在能量转化过程中本身低的导电性,导致其在电催化中的应用受限[41, 42]。除了刚才提到的可以通过原位生长在导电基底上来提高材料暴露的活性位点和导电性外,研究者同时提出了另外的解决办法,就是将LDHs与导电材料杂化,例如碳材料、碳纳米管、碳量子点、石墨烯等等。杂化的方式一般有两种,第一种是通过水热的方法直接将LDHs生长在碳材料上;另外一种是将剥离的带正电的LDH纳米片和带负电的氧化石墨烯分子自组装,形成超分子结构。Gong等[13]首次报道了将NiFe LDH超薄纳米片直接生长在温和氧化的碳纳米管的功能化官能团上形成NiFe LDH/CNTs复合物。整个材料的结构展示了NiFe LDH/CNTs复合物相互交联的纳米导电网络,同时,NiFe LDH纳米片分散在碳纳米管的表面。最终形成的复合物在碱性环境中展现了较高的析氧催化活性和较高的稳定性,而且周转频率也要高于Ni和Fe混合氧化物三倍。作者认为高活性的原因在于NiFe LDH和CNTs在促进电子转移中的相互作用,通过相互交联的纳米网络,借助碳材料极高的导电性,有效的提高了LDH的导电性,促进电子的转移,使LDHs内在的催化活性充分的发挥出来,从而实现了较快的析氧反应速率。文献报道了CQDs和NiFe LDH形成的复合物,其中CQDs尺寸仅有5 nm,具有极高的导电性[43]。CQDs/ NiFe LDH 复合物展现了好的稳定性以及卓越的OER电催化活性。在电流密度为10 mA/cm2下,过电势大约为235 mV, 这个结果要超过之前报道的NiFe 化合物。文献通过引入温和氧化石墨烯和单壁碳纳米管来调节和控制NiFe LDH生长在纳米碳的框架中[39]。氧化的碳结构中的含氧官能团和缺陷可以吸附离子成核,形成尺寸可调的层状氢氧化物,进而形成多级介孔结构,有利于质子电荷的转移和有效的利用活性位点。构建的这种复合物能够实现在电流密度为10 mA/cm2下的过电势为240 mV。文献通过简单的化学沉积(CBD)方法,将CoMn LDH和NiMn LDH生长在碳纳米管的骨架上形成复合物[34]。由于双金属氢氧化物提供的大量的活性位点以及碳基材料理想的导电性,获得的复合材料催化剂相对于高活性的过渡金属化合物,展现了较强的催化活性。对于剥离的LDH的纳米片,虽然活性面积增加了,但纳米片之间的聚集又是不可避免的,为了克服这个问题,研究者考虑到通过带正电的LDH纳米片与带负电的氧化石墨烯分子进行分子间自组装,得到的复合物同时兼具了两个成分之间的优势,既避免了团聚,同时又提升了LDH的稳定性和导电性。此外,通过自组装的方法,Sasaki等[44]成功的将带正电的LDH纳米片与带负电的氧化石墨烯组装在一起形成了一个类似超晶格的结构。在经过热还原处理后,形成的复合物展现了非常好的OER性能,较小的过电势0.23V, 塔菲尔斜率为42 mV/dec。这些卓越的性能主要是由于两种纳米片面对面的界面杂化提升了电子在材料中的转移速率。随后,又进一步将NiMn LDH纳米片与氧化石墨烯絮凝得到超晶格复合物,同样实现了较高的OER活性[45]。 尽管LDHs材料本身具有很好的催化活性,但是由于内在差的导电性和稳定性,使得研究者提出了一些的新的方法,通过不同材料之间的一个结合来改善LDHs导电性和稳定性从而进一步提高LDHs的催化活性。目前文献中主要报道了两种方法:①通过异质元素的掺杂;②通过与其他功能材料杂化形成多级复合材料。通过这两种方式复合的优点在于首先是要利用其他元素的优势来提高LDHs材料的性能,其次是通过两个成分之间的协同作用为电解质的渗透、质子的吸附提供更多的可能。 2.3.1 通过多级生长的方式制备复合材料 目前文献中报道的关于LDHs在电催化中的应用主要集中在析氧反应,而在实际的工业应用中,若能实现在同一电解质中的全解水将会大大的简化装置,减小工业成本,实现资源的最大化利用[46,47]。基于这样的想法,最近,Hou等通过在电化学剥离的石墨片上原位生长非化学计量的CoSe纳米片, 随后再通过水热处理将NiFe LDH沉积在CoSe纳米片上,得到了一个全新的三维多级复合物(如图2)[48]。首先,在这篇文章中用到的基底是导电性极高的石墨基底,并且通过电化学剥离的方法实现了石墨片表面的剥离从而增加了比表面积,更利于吸附和生长功能材料。其次,本文用到的是非化学计量比的Co0.85Se,相较于报道的比较多的CoSe2,这种非化学计量比的Co0.85Se,存在Co 3d和Se 4p 电子轨道的重叠,从而具有内在的半金属特征,导致高的电导性,这种高的电导性成为电催化材料的一个优势。实际上非化学计量比的Co0.85Se具有大量暴露的活性位点,有利于在碱性介质中实现全解水。在这里,OH-1能够通过静电吸附附着在Co0.85Se纳米片的表面。除了修饰形貌,设计一些Co基复合材料,利用不同成分之间的相互作用也能够实现对催化性能的巨大提升。NiFe LDH 由于自身存在的催化活性成为构建这个复合物极好的候选者。Feng等第一次报道了这样的结构,得到的复合物具有较高的比表面积156 m2/g和强的相互作用,实现了优异的双功能催化性能。在OER中, 电流密度为150 mA/cm时只需要过电势1.5 V。 在HER中, 10 mA/cm时的过电势为0.26 V。 更为重要的是当将这种电极材料组装在一个两电极的电解槽中,在20 mA/cm能够实现电压1.71 V, 是能够与Pt/C 和Ir/C催化剂相比较的。 图2 EG/Co0.85Se/NiFe-LDH多级材料合成过程示意图[48]Fig.2 Schematic illustration for the synthesis process of EG/Co0.85Se/NiFe-LDH[48] 最近,本课题组通过两步水热的方法来设计合成了NiCo2O4/NiFe LDH多级结构,具有双功能全解水的特性[49]。通过水热生长将NiCo2O4纳米线生长在酸处理过的泡沫镍上,随后通过第二步水热将NiFe LDH超薄纳米片垂直生长在NiCo2O4纳米线(图3a)。通过对合成的NiCo2O4纳米线的透射电子显微镜的观察发现整个纳米线长度在几个微米左右宽度在200 nm左右。重要的是整个纳米线是由许多小的颗粒拼接在一起,颗粒之间相互连接,彼此又存在一定的距离,这种结构极大地暴露了活性位点,同时具有优良的电子传输特性。通过进一步EDX分析发现在NiCo2O4的表面元素Ni的组成含量偏高说明表面是富Ni层,同时显微镜观察表明NiFe LDH是以该富Ni层为晶核生长的。一方面小晶核颗粒使得随后生长的NiFe LDH纳米片具有超薄的特性,为催化反应提供了大量的活性位点,另一方面两种组成成分NiCo2O4和NiFe LDH通过富Ni层结合,大大增强了两者之间的相互作用。这种新型材料的分级结构特征结合组成成分之间的强相互作用,为电子的传输、电解质的渗透均提供了便利的通道,从而同时实现了卓越的析氢和析氧催化活性 (图3b和3c)。在三电极体系下,1 M KOH 电解质中测试在析氧反应中启动电势为1.46 V, 在电流密度为50 mA/cm2下的过电势仅需要290 mV, 在析氢反应中启动电势为83 mV, 在电流密度为10 mA/cm2下的过电势仅需要192 mV。进一步,用NiCo2O4/NiFe LDH两个电极组装在一个电解槽中实现了双功能的电解水,电解水的电压只需要1.60 V。能够实现如此优越的性能,认为来自以下三个方面:①使用了导电基底而且通过直接生长的方式避免了使用高分子粘接剂;②特殊的多级纳米结构最大限度地暴露了活性位点;③两种组成成分通过富Ni层结合,相互协同作用增强,利于电子和电解质的传输,进而实现最优的催化性能。 图3 NiFe LDH/ NiCo2O4多级结构的合成示意图(a), OER(b)和HER(c)线性循环伏安曲线 [49]Fig.3 Schematic illustration for the construction of the hybrid hierarchical structure(a). LSV curves of OER(b) and HER(c) [49] 2.3.2 通过LDH层间离子交换制备复合材料 LDHs是由带正电的金属片层和带负电的层间阴离子组成的,对于大部分合成的LDHs,层间通常是碳酸根离子,如果需要剥离LDHs形成单片带正电的片层,通常需要交换层间的碳酸根离子,从而实现在甲酰胺中的剥离。考虑到LDH层间的可调节性,研究者们考虑是否可以通过交换的方式,将其他的离子交换到LDHs的层间从而来提升催化性能。Long等[50]报道了通过交换的方式将氧化石墨烯交换到FeNi LDHs的层间,形成一种超晶格的结构。来自LDHs的催化活性和来自石墨烯的增强电子转移的协同作用,使得FeNi-GO表现出良好的催化性能。在这里,FeNi LDH阳离子层是能够从FeNi LDH剥离的,具有良好的电催化析氧活性。然后将功能化的氧化石墨烯与可交换的FeNi LDH进行搅拌,实现功能化的石墨烯插入到NiFe LDH的层间(如图4a)。这种高的催化活性主要是由于内在的FeNi LDH的活性引起的,而且,GO和FeNi LDH之间强的相互作用,能够暴露催化活性位点,提升电荷转移,从而实现OER表现的优化。 当层间是GO, 能够实现过电势0.21 V和塔菲尔斜率仅有40 mV/dec。如果进一步的将层间的GO还原为rGO, 过电势能够进一步降低到0.195 V, 且在过电势为0.3 V时的周转频率为0.98 /s (如图4b和4c)。 随后,文献探究了不同尺寸阴离子的插入LDHs层间对析氧反应的影响,发现层间距增加会导致OER活性的增加,可能原因是更大的层间距暴露了更多的活性位点,同时降低了电荷转移电阻[21]。另外Han等[51]在钼酸根溶液中直接合成钼酸根插入的NiFe LDH。形成的超薄NiFe LDH纳米片性能是规则NiFe LDH纳米片的3倍。 过渡金属硒化物,氮化物,硫化物具有较好的析氢性能,通常合成他们的方法是在管式炉中对过渡金属氧化物或者氢氧化物通入相应的气体来得到[52-57]。尽管在这方面的研究报道很多,但仍然存在许多的问题,比如,这些催化剂导电性较差,而且限制了活性位点的暴露,严重影响了析氢反应的表现。最近,研究者们对于提升析氢催化性能提出了两种有效策略。①通过将催化剂与导电网络结合在一起;②构建特殊的结构,比如中空、介孔或者多级结构。基于以上观点,人们试图通过一些方法将LDH转为其他功能性的化合物从而实现LDHs析氧性能的提升以及获得较优的析氢性能。2015年,Jia等[58] 图4 GO/FeNi LDH 合成示意图 (a), 合成的电极材料的线性循环伏安曲线(b)和塔菲尔曲线(c) [50]Fig.4 Scheme for the synthesis of FeNi LDH/GO hybrid (a), LSV curves(b) and Tafel slopes(c) of OER catalyzed by the as-prepared catalysts[50] 报道了通过选择性腐蚀NiGa LDH转化为介孔氢氧化物,并进一步通过溶液法转化为硒化镍。由此获得的β-Ni(OH)2和硒化镍具有高的析氧和析氢活性,同时证明了具有丰富活性位点和高比表面积的介孔纳米结构的应用。在酸性和碱性环境中,硒化镍都展现了高的析氢活性,低的过电势,小的塔菲尔斜率和高的稳定性。同时将两种材料作为全解水的电极获得了较低的电压。Jia等[59]报道了从反向微乳液方法合成的超薄NiFe LDH在热氨解的条件下获得Ni3FeN纳米颗粒。这种纳米颗粒展现了非常高的OER和HER活性,在10 mA/cm下过电势为158和280 mV, 塔菲尔斜率分别为42和46 mV/dec。这种催化活性提高的主要是由高的活性位点和高的电导性导致的。Ma等[60]报道了硫化生长在泡沫镍上的NiCo LDH, 获得的NiCo2S4/NF具有超薄介孔结构,能够提供大量的暴露的活性位点,同时高导电性的泡沫镍能够促进电子的转移 (如图5)。三维网络结构也能够促进电解质的扩散和渗透。电化学测试也揭示了NiCo2S4/NF在碱性环境中析氢性能的提升(过电势17 mV, 塔菲尔斜率为84.5 mV/dec)。最近,在文献中报道了水热生长NiCo LDH在碳布上,后通过磷化得到NiCoP,实现了双功能催化。在电流密度为50 mA/cm2时,仅需要1.77 V的电压。优异的性能主要归因于电极材料内在的催化活性,快速的电子转移和独特的亲水结构。 图5 三维网络NiCo LDH硫化的示意图[60]Fig.5 Schematic illustration of the 3D-networked NiCo LDH/Ni foam sulfidation process [60] 由于具有易于合成、结构可调和相对较低的成本等优点,使得LDHs成为一类理想的电极材料。目前,不同种类的LDHs和LDH基复合材料已经成功地应用在能量转化、储存以及催化领域。基于片层内阳离子的可调性以及层间插入阴离子的可交换性,已经合理地设计和合成出一系列LDH基材料。本文试图归纳了近期LDH基电极材料在电催化水解中的研究进展。许多不同研究成果已经证明其在催化领域具有重要的应用潜力,因此有理由相信在不远的未来人们将能够通过这类廉价而又丰富的材料实现催化水解和可清洁能源再生。尽管目前在设计和合成LDH基材料方面已经取得了一些突破性的进展,但是由于LDH基材料本身存在的一些缺陷(酸性和中性电解质中稳定性较差,电子传输速率有限等),仍然需要进一步的研究,主要集中在以下三个方面: (1)设计和合成高质量低成本的LDH基材料以应用于电催化反应,并使合成过程更加绿色化和安全化;其中包括控制LDH的成分组成、尺寸以及形貌,尤其是一些具有较大比表面积的结构,例如中空结构,核壳结构等。 (2)基于LDH基材料优越的层间化学反应特性,可以通过引入一些功能性的基团,如钼酸根、氯铂酸根、铁氰酸根等,进而得到一些新型的LDH基复合材料。 (3)考虑到LDH基材料的催化反应多局限于析氧半反应中,因此设计更多与LDH基复合的无机功能材料,使其同时具有优越的析氢反应特性,从而实现全解水功能,有利于降低电解水设备的成本,同时也将为电解水的工业应用提供更大的可能性。 References [1] Wang J, Cui W, Liu Q.etal.AdvancedMaterials[J], 2016, 28(2): 215-30. [2] Zou X, Zhang Y.ChemicalSocietyReviews[J], 2015, 44(15): 5148-80. [3] Mallouk T E.NatureChemistry[J], 2013, 5(5): 362-3. [4] Joya K S, Joya Y F, Ocakoglu K,etal.AngewandteChemieInternationalEdition[J], 2013, 52(40): 10426-37. [5] Kong D, Cha J J, Wang H,etal.Energy&EnvironmentalScience[J], 2013, 6(12): 3553-8. [6] Zou X, Goswami A, Asefa T.JournaloftheAmericaChemicalSociety[J], 2013, 135(46): 17242-5. [7] Esposito D V, Hunt S T, Stottlemyer A L,etal.AngewandteChemieInternationalEdition[J], 2010, 49(51): 9859-62. [8] Hu T C, Wen C.JournaloftheElectrochemicalSociety[J], 1992, 139(2): 158-62. [9] Zheng Y, Jiao Y, Jaroniec M,etal.AngewandteChemieInternationalEdition[J], 2015, 54(1): 52-65. [10] Wang X, Li W, Xiong D,etal.AdvancedFunctionalMaterials[J], 2016, 26(40): 67-77. [11] Jin Y, Wang H, Li J,etal.AdvancedMaterials[J], 2016, 28(19): 3785-90. [12] Liu X, Zheng H, Sun Z,etal.ACSCatalysis[J], 2015, 5(3): 1530-8. [13] Gong M, Li Y, Wang H,etal.JournaloftheAmericanChemicalSociety[J], 2013, 135(23): 8452-5. [14] Long X, Wang Z, Xiao S,etal.MaterialsToday[J], 2016, 19(4): 213-26. [15] Qian L, Lu Z, Xu T,etal.AdvancedEnergyMaterials[J], 2015, 5(13): 1500245. [16] Wang L, Huang X, Xue J.ChemSusChem[J], 2016, 9(14): 1835-42. [17] Fan K, Chen H, Ji Y,etal.NatureCommunications[J], 2016, 7(11): 981-8. [18] Abellán G, Carrasco J A, Coronado E,etal.JournalofMaterialsChemistryC[J], 2014, 2(19): 3723-9. [19] Jiang J, Zhang A, Li L,etal.JournalofPowerSources[J], 2015, 278(4): 45-51. [20] Xing J, Wu S, Ng K, Simon Y.RSCAdvances[J], 2015, 5(108): 88780-6. [21] Xu Y, Hao Y, Zhang G,etal.RSCAdvances[J], 2015, 5(68): 55131-5. [22] Li L, Ma R Z, Ebina Y,etal.ChemistryofMaterials[J], 2005, 17(17): 4386-91. [23] Liu Z P, Ma R Z, Osada M,etal.JournaloftheAmericanChemicalSociety[J], 2005, 127(40): 13869-74. [24] Li L, Sasaki T,etal.ChemistryofMaterials[J], 2005, 17(17): 4386-4391. [25] Fan G, Li F, Evans D G,ChemicalSocietyReviews[J], 2014, 43(20): 7040-66. [26] Yin H, Tang Z.ChemicalSocietyReviews[J], 2016, 24(16): 131-154. [27] Iyi N, Ebina Y, Sasaki T.JournalofMaterialsChemistry[J], 2011, 21(22): 8085. [28] Liu Z P, Ma R Z, Osada M,etal.JournalofTheChemicalSociety[J], 2006, 128(14): 4872-80. [29] Ma R, Liu Z, Takada K,etal.JournaloftheAmericanChemicalSociety[J], 2007, 129(16): 5257-63. [30] Li Y, Zhang L, Xiang X,etal.JournalofMaterialsChemistryA[J], 2014, 2(33): 13250. [31] Shao M, Zhang R, Li Z,etal.ChemicalCommunications[J], 2015, 51(88): 15880-93. [32] Yan K, Wu G, Jin W.EnergyTechnology[J], 2016, 4(3): 354-68. [33] Tang C, Wang H S, Wang H F,etal.AdvancedMaterials[J], 2015, 7(13): 527-34. [34] Jia G, Hu Y, Qian Q,etal.ACSAppliedMaterials&Interfaces[J], 2016, 8(23): 14527-34. [35] Han X, Yu C, Yang J,etal.AdvancedMaterialsInterfaces[J], 2016, 3(7): 1500782. [36] Song F, Hu X.JournaloftheAmericanChemicalSociety[J], 2014, 136(47): 16481-4. [37] Song F, Hu X.NatureCommunications[J], 2014, 5(4): 477-82. [38] Chen S, Duan J, Jaroniec M,etal.AngewandteChemieInternationalEdition[J], 2013, 52(51): 13567-70. [39] Zhu X, Tang C, Wang H,etal.JournalofMaterialsChemistryA[J], 2015, 3(48): 24540-6. [40] Luo J, Im J H, Mayer M T,etal.Science[J], 2014, 345(6204): 1593-6. [41] Liang H, Meng F, Caban A M.,etal.NanoLetters[J], 2015, 15(2): 1421-7. [42] Yang Q, Li T, Lu Z,etal.Nanoscale[J], 2014, 6(20): 11789-94. [43] Tang D, Liu J, Wu X,etal.ACSAppliedMaterials&Interfaces[J], 2014, 6(10): 7918-25. [44] Sasaki T.ACSNano[J], 2015, 9(1): 977-82. [45] Ma W, Ma R, Wu J,etal.Nanoscale[J], 2016, 8(19): 10425-32. [46] Yin J, Zhou P, An L,etal.Nanoscale[J], 2016, 8(3): 1390-400. [47] Li Y, Zhang H, Jiang M,etal.NanoResearch[J], 2016, 17(23): 5227-34. [48] Hou Y, Lohe M R., Zhang J,etal.Energy&EnvironmentalScience[J], 2016, 9(2): 478-83. [49] Wang Z, Liu W, Geng F,etal.AppliedMaterials&Interfaces[J], 2017, 9(2): 1488-95. [50] Long X, Li J, Xiao S,etal.AngewandteChemieInternationalEdition[J], 2014, 53(29): 7584-8. [51] Han N, Zhao F, Li Y.JournalofMaterialsChemistryA[J], 2015, 3(31): 16348-53. [52] Zhang M, de Respinis M, Frei H.NatureChemistry[J], 2014, 6(4): 362-7. [53] Zhao Z, Wu H, He H,etal.AdvancedFunctionalMaterials[J], 2014, 24(29): 4698-705. [54] Zhu X, Tang C, Wang H,etal.JournalofMaterialsChemistryA[J], 2016, 4(7): 245-50. [55] Feng L L, Yu G, Wu Y,etal.JournalofTheAmericanChemicalSociety[J], 2015, 137(44): 14023-6. [56] Xu R, Wu R, Shi Y,etal.NanoEnergy[J], 2016, 24(10): 3-10. [57] Cui W, Cheng N, Liu Q,etal.ACSCatalysis[J], 2014, 4(8): 2658-61. [58] Liang H, Li L, Meng F,etal.ChemistryofMaterials[J], 2015, 27(16): 5702-11. [59] Jia X, Zhao Y, Chen G,etal.AdvancedEnergyMaterials[J], 2016, 6(10): 1502585. [60] Ma L, Hu Y, Chen R,etal.NanoEnergy[J], 2016, 24(1): 39-47. (编辑 吴 琛) Progress in Research of Transition Metal Layered Double Hydroxide Electrocatalysis WANG Zhiqiang,LIU Weihong,GENG Fengxia (College of Chemistry, Chemical Engineering and Materials Science, Soochow University, Suzhou 215123, China) TQ116.2+1 A 1674-3962(2017)09-0659-08 2016-09-15 国家自然科学基金资助项目(51402204) 王志强,男,1990年生,硕士研究生 耿凤霞,女,1980年生,教授,博士生导师,Email: gengfx@suda.edu.cn 10.7502/j.issn.1674-3962.2017.09.082 LDHs在电催化水解中的应用
2.1 单一的LDHs
2.2 改善LDH导电性提升催化性能

2.3 LDH与其他功能性无机材料的复合

2.4 LDH转化为其他功能材料


3 结 语
猜你喜欢
油气田地面工程(2022年8期)2022-10-02
防爆电机(2020年4期)2020-12-14
装备维修技术(2020年5期)2020-11-20
矿产综合利用(2020年1期)2020-07-24
河北理科教学研究(2020年1期)2020-07-24
化工管理(2020年16期)2020-01-14
分析化学(2018年12期)2018-01-22
分析化学(2018年1期)2018-01-18
分析化学(2017年9期)2017-10-16
中小企业管理与科技·下旬刊(2016年12期)2017-01-17
