
UPLC-MS/MS法同时检测抗风湿中药制剂中非法添加的12种非甾体抗炎药
2017-10-13 08:30:04言慧洁刘伟夏青松潘源虎湖北省直属机关医院武汉43007华中农业大学动物科技与动物医学院武汉43007
中国药房 2017年27期
言慧洁,刘伟,夏青松,潘源虎(.湖北省直属机关医院,武汉43007;.华中农业大学动物科技与动物医学院,武汉 43007)
UPLC-MS/MS法同时检测抗风湿中药制剂中非法添加的12种非甾体抗炎药
言慧洁1*,刘伟1,夏青松1,潘源虎2(1.湖北省直属机关医院,武汉430071;2.华中农业大学动物科技与动物医学院,武汉 430072)
目的:建立同时检测抗风湿中药制剂中非法添加的12种非甾体抗炎药。方法:采用超高效液相色谱-质谱法。色谱条件:色谱柱为Hypersil Golden C18,流动相为5 mmol/L甲酸铵溶液-甲醇(梯度洗脱),流速为0.2 mL/min,柱温为40℃,进样量为2 μL。质谱条件:离子源为电喷雾离子源,气帘气压为25 kPa,雾化气压为60 kPa,辅助气压为55 kPa,电喷雾电压为4 500 V,离子源温度为650℃,采集方式为多反应监测模式。结果:对乙酰氨基酚、乙酰水杨酸、氨基比林、美洛昔康、布洛芬、萘普生、舒林酸、尼美舒利、双氯芬酸、吲哚美辛、酮洛芬、塞来昔布检测质量浓度线性范围分别为0.01~2.0 μg/mL(r=0.995 6)、0.05~5.0 μg/mL(r=0.997 6)、0.01~2.0 μg/mL(r=0.998 7)、0.02~5.0 μg/mL(r=0.995 0)、0.02~5.0 μg/mL(r=0.995 3)、0.02~5.0 μg/mL(r=0.996 5)、0.05~5.0 μg/mL(r=0.995 4)、0.02~5.0 μg/mL(r=0.996 0)、0.05~5.0 μg/mL(r=0.995 9)、0.02~5.0 μg/mL(r=0.995 7)、0.02~5.0 μg/mL(r=0.996 8)、0.01~2.0 μg/mL(r=0.998 7);定量限≤0.20 mg/g,检测限≤0.05 mg/g;精密度、稳定性、重复性试验的RSD<5.0%;加样回收率为80.8%~114.2%(RSD为3.85%~7.32%,n=9)。结论:该方法操作简便,精密度、稳定性、重复性好,可用于抗风湿中药制剂中非法添加的12种非甾体抗炎药的同时检测。
非甾体类抗炎药;中药制剂;非法添加;超高效液相色谱-质谱法
ABSTRACTOBJECTIVE:To develop a method for simultaneous determination of 12 nonsteroidal anti-inflammatory drugs(NSAIDs)illegally added into antirheumatic TCM preparations.METHODS:UPLC-MS/MS was adopted.The determination was performed on Hypersil Golden C18column with mobile phase consisted of 5 mmol/L ammonium formate solution-methanol(gradient elution)at the flow rate of 0.2 mL/min.The column temperature was 40 ℃,and the sample size was 2 μL.A tandem quadrupole mass spectrometer equipped with electrospray ionization source was used in positive-negative ion mode:curtain gas of 25 kPa,atomizing gas of 60 kPa,auxiliary gas of 55 kPa,electrospray voltage of 4 500 V,ion source temperature of 650℃.The multiple reaction monitoring mode was performed.RESULTS:The linear ranges of acetaminophen,acetylsalicylic acid,aminopyrine,meloxicam,ibuprofen,naproxen,lam acid,nimesulide,diclofenac,indomethacin,ketoprofen and celecoxib were 0.01-2.0 μg/mL(r=0.995 6),0.05-5.0 μg/mL(r=0.997 6),0.01-2.0 μg/mL(r=0.998 7),0.02-5.0 μg/mL(r=0.995 0),0.02-5.0 μg/mL(r=0.995 3),0.02-5.0 μg/mL(r=0.996 5),0.05-5.0 μg/mL(r=0.995 4),0.02-5.0 μg/mL(r=0.996 0),0.05-5.0 μg/mL(r=0.995 9),0.02-5.0 μg/mL(r=0.995 7),0.02-5.0 μg/mL(r=0.996 8),0.01-2.0 μg/mL(r=0.998 7),respectively.The limits of quantitation were no more than 0.20 mg/g,and the limits of detection were no more than 0.05 mg/g.RSDs of precision,stability and reproducibility tests were all lower than 5.0%.The recoveries were 80.8%-114.2%(RSD was 3.85%-7.32%,n=9).CONCLUSIONS:The established method is simple,accurate,stable and reproducible,and can be used for simultaneous determination of 12 NSAIDs illegally added into antirheumatic TCM preparations.
KEYWORDSNonsteroidal anti-inflammatory drugs;TCM preparation;Illegally added;UPLC-MS/MS
非甾体类抗炎药(NSAIDs)是临床上常用的解热镇痛抗炎药,机制为通过抑制体内环氧化酶(COX)活性而减少局部组织前列腺素(PG)的生物合成,除具有解热、镇痛作用外,多数还有抗炎、抗风湿的作用,也是治疗类风湿关节炎及强直性脊柱炎的首选药物[1]。但是使用不当或长期过量的应用亦会导致消化道溃疡、肝肾毒性和心血管系统疾病[2-3]。2004年德国默克公司生产的罗非昔布(商品名:万络)退市风波更是让人们重新审视选择性COX-2抑制剂这类NSAIDs可能引发的心血管事件[4]。中药制剂可以改善风湿性疾病症状,控制病情进展,阻止不可逆的骨质破坏,从而改善患者生活质量,在风湿和类风湿关节炎的治疗方面具有独特的优势[5]。但为了快速起效或增强药效,违法添加NSAIDs的现象时有发生,长期服用会给患者的身体健康带来严重威胁。近年来,许多文献报道了NSAIDs的检测方法如薄层色谱法[6]、高效液相色谱法[7-8]、高效液相色谱-质谱法等(HPLC-MS)[9-10]。本研究建立了超高效液相色谱-串联质谱法(UPLC-MS/MS)同时测定抗风湿类中药制剂中可能非法添加的12种NSAIDs的方法,以期为其质量控制提供可靠的技术支持。
1 材料
1.1 仪器
Triple-Quadrupole Instrument型UPLC-MS/MS仪(美国应用生物系统公司);AUW-3200D型电子分析天平(日本Shimadzu公司,感量:0.01 mg);KQ3200型超声波清洗仪(昆山市超声仪器有限公司)。
1.2 药品与试剂
试验所用样品均为市售抗风湿丸剂、片剂、胶囊剂、(编号:1~10);对乙酰氨基酚(AMP)对照品(批号:100018-201409)、乙酰水杨酸(ASP)对照品(批号:100113-201104)、氨基比林(ATD)对照品(批号:100503-201302)、美洛昔康(MXC)对照品(批号:100679-201102)、萘普生(NPX)对照品(批号:100198-201205)、舒林酸(SLD)对照品(批号:100335-200001)、尼美舒利(NSL)对照品(批号:100555-201202)、双氯芬酸(DCF)对照品(批号:100334-200302)、布洛芬(IBF)对照品(批号:100179-201105)、吲哚美辛(IMC)对照品(批号:100258-200904)、酮洛芬(KPF)对照品(批号:100337-201104)均购自中国食品药品检定研究院;塞来昔布(CLB)对照品(美国Sigma公司,批号:LOT F0L037),以上对照品纯度均>99%;甲醇为色谱纯,其余试剂均为分析纯,水为超纯水(电导率:18.2 MΩ/cm)。
2 方法与结果
2.1 试验条件
2.1.1 色谱条件 色谱柱:Hypersil Golden C18(150 mm×2.1 mm,5 μm);流动相:5 mmol/L甲酸铵溶液(A,pH4)-甲醇(B),梯度洗脱(0~5 min,90%A;5~15 min,90%→60%A;15~25 min,60%→20%A;25~30 min,20%→90%A);流速:0.2 mL/min;柱温:40 ℃;进样量:2 μL。
2.1.2 质谱条件 离子源:电喷雾离子源(ESI);气帘气压(CUR):25 kPa;雾化气压(GAS1):60 kPa;辅助气压(GAS2):55 kPa;电喷雾电压:4 500 V;离子源温度:650℃;采集方式:多反应监测(MRM)模式。
2.2 溶液的制备
2.2.1 对照品溶液 精密称取各待测成分对照品25.0 mg,分别置于25 mL量瓶中,加甲醇溶解并定容,制成AMP、ASP、ATD、MXC、NPX、SLD、NSL、DCF、IBF、IMC、KPF、CLB质量浓度均为1 mg/mL的单一对照品溶液,于-20℃下贮藏。
2.2.2 供试品溶液 取样品0.5 g(片、丸研磨成细粉,胶囊取内容物研细),置于100 mL具塞锥形瓶中,精密加甲醇40 mL,超声(功率:250 W,频率:40 kHz)提取20 min,静置后倾出提取液,残渣用甲醇重复提取1次,合并提取液并定容至100 mL。取上述溶液1 mL,加甲醇定容至100 mL,经0.22 μm微孔滤膜滤过,取续滤液,即得。
2.2.3 空白对照溶液 以甲醇作空白对照溶液。
2.3 专属性试验
取“2.2.1”项下单一对照品溶液适量,按“2.1”项下试验条件进样测定,记录色谱,详见图1。结果,AMP、ASP、ATD、MXC、NPX、SLD、NSL、DCF、IBF、IMC、KPF、CLB 保留时间分别为 4.12、7.43、16.62、14.70、16.37、21.02、16.30、24.45、16.85、21.43、20.77、20.24 min,表明各待测成分分离度良好。
2.4 12种化学物质的质谱检测参数
将单个化学物质的对照品溶液进行分析,得到目标化合物的质荷比。为了进一步对化学物质进行确证分析,通过分析单个化学物质对照品溶液,优化去簇电压和碰撞电压,以灵敏度高、稳定性好作为离子化模式选择和提取离子选择依据(详见表1,表中“a”为定量离子),其中ASP、MXC、IBF、NPX、NSL和DCF使用ESI(-)模式,其余6个化学物质使用ESI(+)模式。
2.5 线性关系考察
分别精密量取“2.2.1”项下单一对照品溶液各适量,分别置于10 mL量瓶中,加甲醇定容,制成系列对照品溶液。精密量取上述系列对照品溶液各2 μL,按“2.1”项下试验条件进样测定,记录峰面积。以各待测成分质量浓度(x,μg/mL)为横坐标、峰面积(y)为纵坐标进行线性回归,得回归方程与线性关系,详见表2。
2.6 定量限与检测限考察
分别精密量取“2.2.1”项下单一对照品溶液适量,倍比稀释,并按“2.1”项下试验条件进样测定,当信噪比为10∶1时,得定量限;当信噪比为3∶1时,得检测限,详见表2。
2.7 精密度试验
取“2.2.1”项下单一对照品溶液各适量,按“2.1”项下试验条件连续进样测定6次,记录峰面积。结果,AMP、ASP、ATD、MXC、NPX、SLD、NSL、DCF、IBF、IMC、KPF、CLB峰面积的RSD<4.15%(n=6),表明仪器精密度良好。
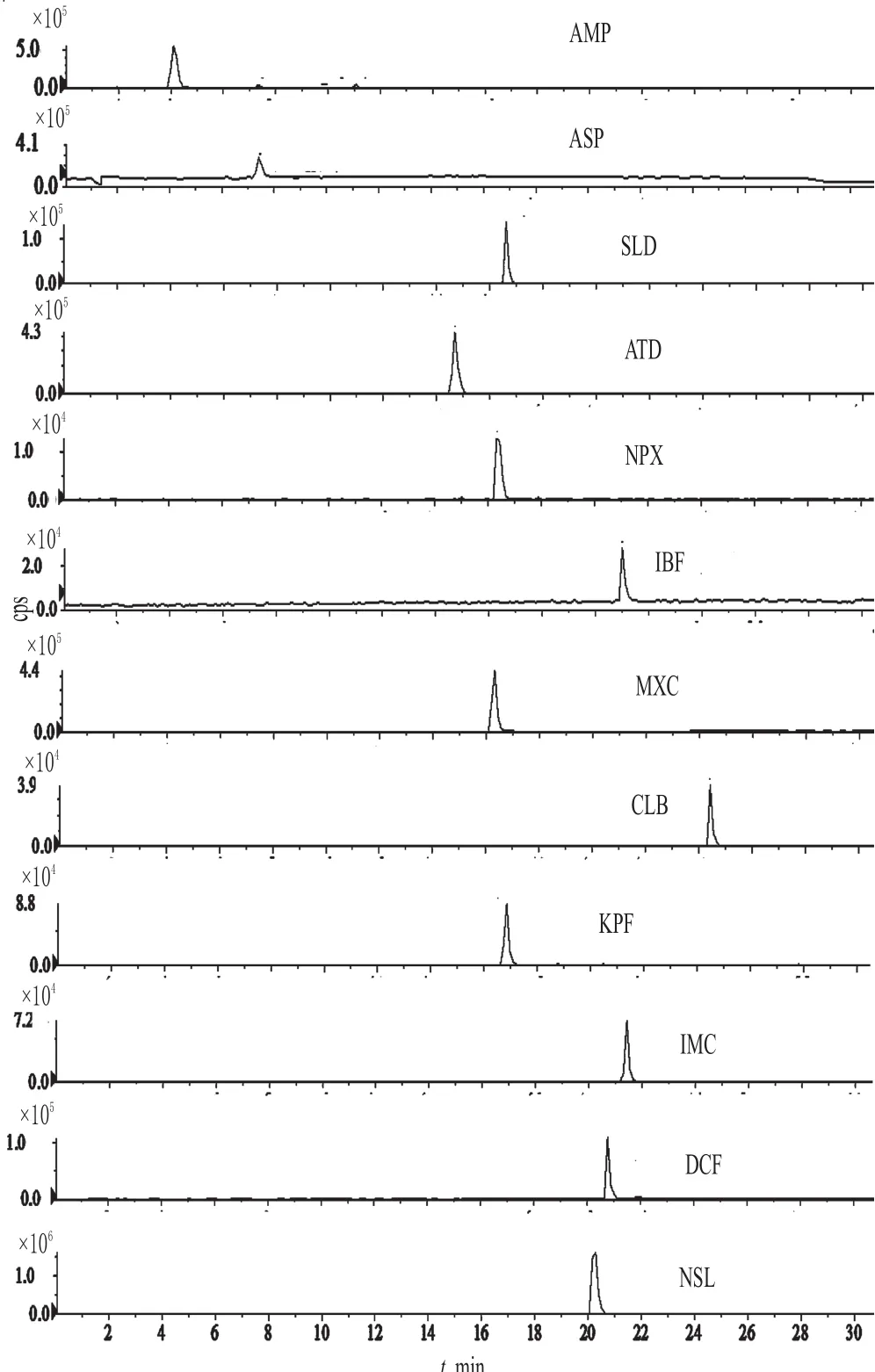
图1 提取离子流图Fig 1 Extraltion ion chromatograms
2.8 稳定性试验
取“2.2.2”项下供试品溶液(编号:2)适量,分别于4 ℃冰箱中放置0、1、3、5、7 d时按“2.1”项下试验条件进样测定,记录峰面积。结果,AMP、ASP、ATD、MXC、NPX、SLD、NSL、DCF、IBF、IMC、KPF、CLB峰面积的RSD<1.21%(n=5),表明供试品溶液4℃下放置7 d内基本稳定。
2.9 重复性试验
精密称取同一批样品(编号:2)适量,按“2.2.2”项下方法制备供试品溶液,共6份,再按“2.1”项下试验条件进样测定,记录峰面积。结果,AMP、ASP、ATD、MXC、NPX、SLD、NSL、DCF、IBF、IMC、KPF、CLB峰面积的RSD<4.69%(n=6),表明本方法重复性良好。
2.10 加样回收率试验
取已知含量样品(编号:2)适量,共9份,分别加入高、中、低质量的待测成分对照品,按“2.2.2”项下方法制备供试品溶液,再按“2.1”项下试验条件进样测定,记录峰面积并计算加样回收率,结果见表3。

表1 质谱检测参数Tab 1 Mass spectrometric parameters
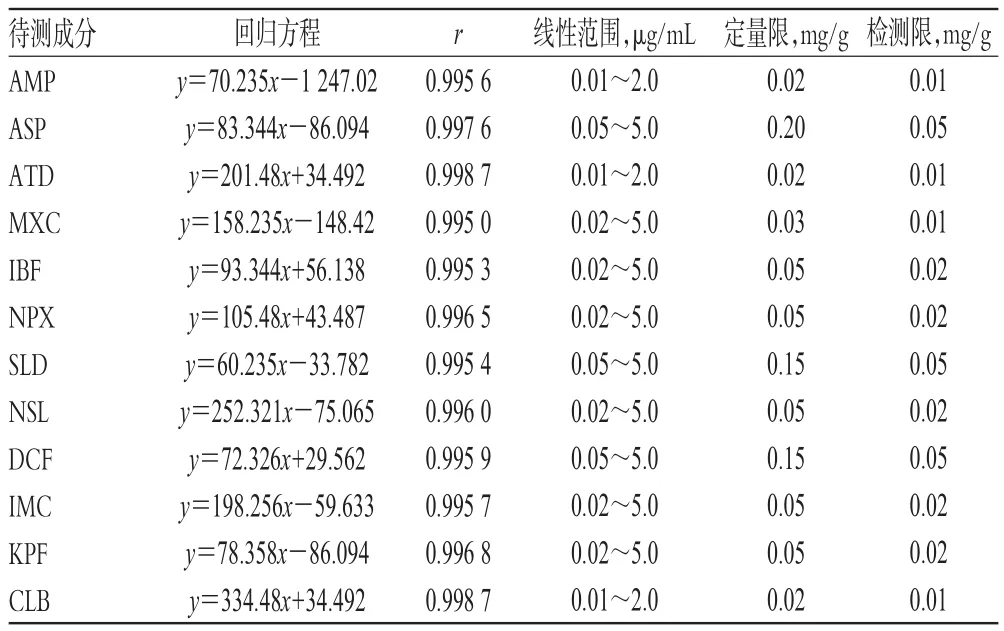
表2 回归方程、线性范围与定量限、检测限Tab 2 Linear regression equations, linear ranges,LO-D and LOQ
2.11 样品中非甾体抗炎药检测
取10批样品各适量,分别按“2.2.2”项下方法制备供试品溶液,再按“2.1”项下试验条件进样测定,记录峰面积并计算样品含量,结果见表4(表中“-”为未检出)。结果,10批样品中有4批样品检测出NSAIDs共有4种,含量范围在7.5~105.2 mg/g,其中1份样品中检出布洛芬、3份样品中检出萘普生、2份样品中检出双氯芬酸、1份样品中检出美洛昔康。
3 讨论
3.1 色谱条件的优化
本试验分别考察了甲醇和乙腈作为流动相B对各色谱峰的峰形和分离度的影响,并且分别添加不同浓度的甲酸或醋酸铵于流动相A中,结果发现以甲醇为流动相B、5 mmol/L醋酸铵水溶液为A相时,各个特征离子色谱图的峰形和分离度良好,采用梯度洗脱可以在30 min内完成12个组分的分离。
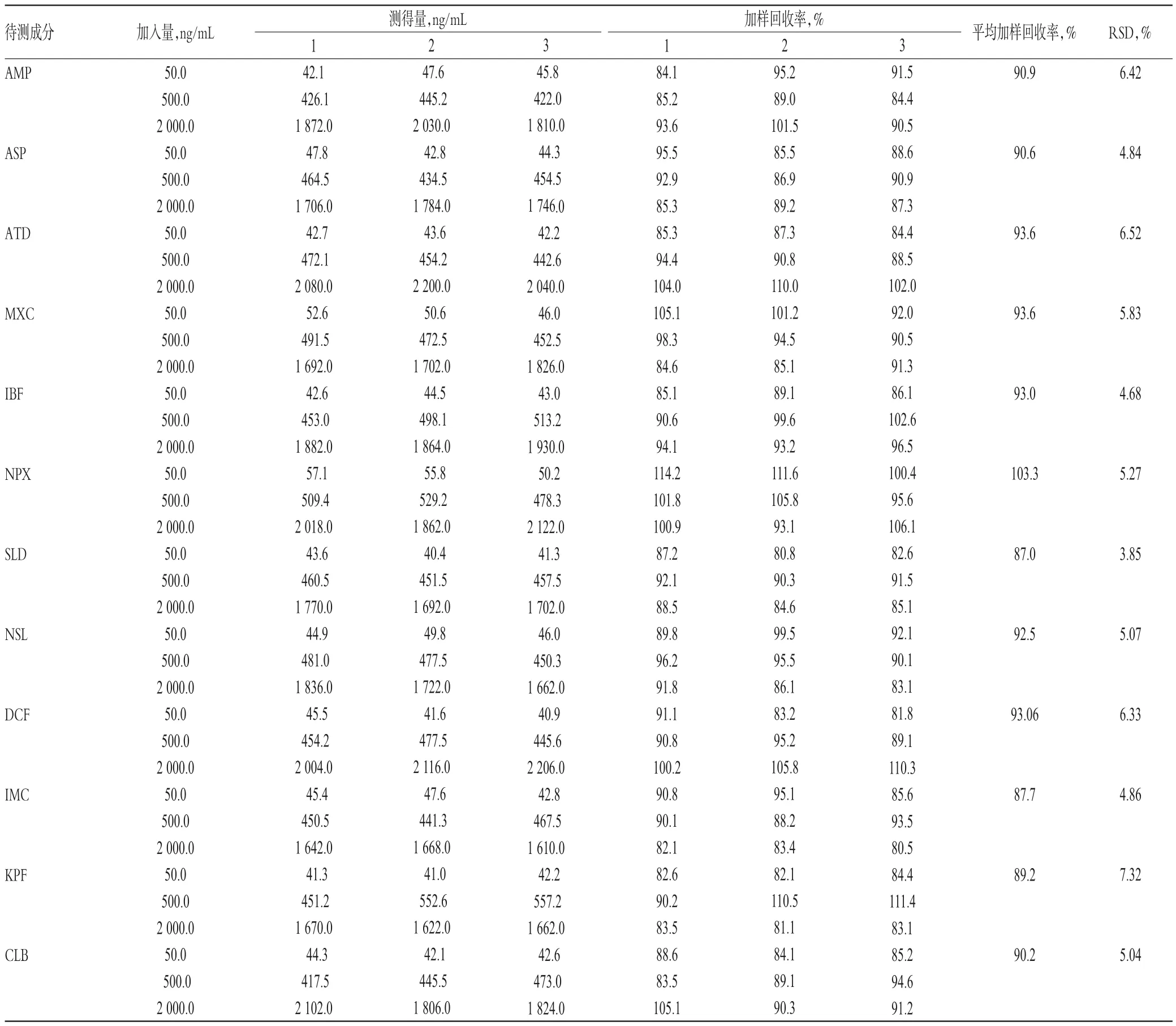
表3 加样回收率试验结果(n=9)Tab 3 Results of recovery tests(n=9)
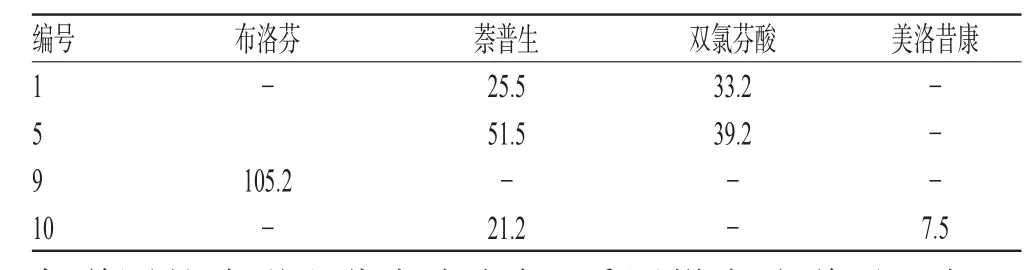
表4 样品中NSAIDs检测结果(n=3,mg/g)Tab 4 Results of NSAIDs in samples(n=3,mg/g)
3.2 提取方法的选择
本研究分别以甲醇、50%甲醇及乙腈作为样品提取溶剂。结果表明,乙腈溶解样品的提取率不及甲醇;选用50%甲醇作为提取溶剂时对AMD、ASP、ATD、SLD和DCF的提取效率较高,但是对其他化学物质较低;采用甲醇作为提取溶剂提取2次时,结果显示对所有化学物质的回收率都达到80%以上;同时,考察结果显示,采用超声提取20 min即可使回收率满足要求,同时基质干扰小。
3.3 液质方法的选择
NSAIDs非法添加在中药制剂中往往干扰成分很多,因此对分析方法的选择性提出了更高的要求。笔者所采用的UPLC-MS/MS法,通过一级全扫描模式获得化合物的准分子离子峰,再提取该准分子离子为母离子进行二级全扫描,获得化学物质的碎片信息。在该模式下确定化学物质的裂解方式之后,提取稳定的碎片离子进行MRM模式检测。即对提取的母离子给予特定能量碰撞后产生碎片离子再次经过提取,从而获得特定的碎片离子峰,这样可以将一些即使在液相色谱系统中无法完全分离的物质,也可通过对其母离子和碎片离子的2次提取,使其质谱行为的差异达到完全分离,不仅提高了特异性,而且还减少了基质干扰,提高了检测的灵敏度。本研究同时采用正负离子扫描方式进行检测,其中AMD、MXC、IBF、NPX、SLD和NCF在正离子方式下不易产生稳定的碎片,所以采用ESI(-)模式,而其余6个化学物质使用ESI(+)模式。
综上所述,本研究建立了抗风湿中药制剂中可能非法添加的12种NSAIDs的UPLC-MS/MS检测方法,并用于实际样品的检测。本方法样品处理简单,没有基质干扰,精密度和准确度高,可作为抗风湿中药制剂或保健品中非法添加抗炎镇痛类化学药成分的监督检测。
[1]郭胜才,潘锋君.非甾体抗炎药的进展[J].华西药学杂志,2000,15(6):469.
[2]甄细娥,王宽,宗鸣.降低非甾体抗炎药致胃肠道不良反应的方法研究进展[J].中国药房,2012,23(6):935-938.
[3]刘德鼎,林博川.非甾体抗炎药治疗类风湿关节炎及对心血管不良影响[J].药学实践杂志,2007,25(5):283-286.
[4]王楠,毛璐.选择性COX-2抑制剂引发心血管事件研究进展[J].中国药物警戒,2012,9(10):625-627.
[5]张靖,周彬,王彦丽,等.抗类风湿性关节炎中药的研究进展[J].中草药,2013,44(15):2189-2194.
[6]李惠敏.薄层色谱法快筛抗风湿类中药制剂和保健食品中4种解热镇痛成分[J].中国现代药物应用,2010,4(8):16-17.
[7]刘志辉.HPLC法同时测定抗风湿类中成药及保健品中非法添加9种解热镇痛类化学药物[J].中国药师,2015,18(7):1116-1119.
[8]宁素云,郭兴杰,张虹,等.HPLC法同时检测清热解毒类中成药中非法添加的9种化学药品[J].中国药事,2009,23(9):907-910.
[9]李丹,文红梅,崔福春,等.LC-MS/MS法快速测定中成药、保健食品中非法添加的36种化学成分[J].药物分析杂志,2010,30(8):1527-1532.
[10]潘炜,顾鑫荣,刘志璋.LC-MS/MS法测定中成药制剂中23个非甾体抗炎药[J].药物分析杂志,2012,32(2):261-266.
(编辑:张 静)
Simultaneous Determination of 12 Nonsteroidal Anti-inflammatory Drugs Illegally Added into Antirheumatic TCM Preparations by UPLC-MS/MS
YAN Huijie1,LIU Wei1,XIA Qingsong1,PAN Yuanhu2(1.Hubei Zhishu Jiguan Hospital,Wuhan 430071,China;2.College of Animal Sciences Technology&College of Veterinary Medicine,Huazhong Agricultural University,Wuhan 430072,China)
R927.2;R917
A
1001-0408(2017)27-3871-05
2016-10-25
2017-04-13)
*副主任药师。研究方向:临床药学。电话:027-87234240。E-mail:butterfly20058@hotmail.com
DOI 10.6039/j.issn.1001-0408.2017.27.36
猜你喜欢
科学导报(2019年19期)2019-09-23 09:04:24
中老年保健(2019年3期)2019-03-04 06:23:42
中国现代药物应用(2017年23期)2017-12-16 07:26:46
当代临床医刊(2017年1期)2017-03-14 06:09:31
中国民族民间医药·上半月(2016年10期)2016-11-19 11:29:23
兽医导刊(2016年18期)2016-04-05 04:43:08
中国医药科学(2015年15期)2015-02-27 12:32:32
健康之路(医药研究)(2014年3期)2014-04-29 13:31:38
中成药(2014年9期)2014-02-28 22:28:54
中国医药指南(2014年23期)2014-01-25 04:52:38
