
胎儿单基因遗传病的无创检测技术研究进展
2017-09-29 02:42周祎蒋馥蔓王子莲
中国产前诊断杂志(电子版) 2017年3期
周祎 蒋馥蔓 王子莲
(中山大学附属第一医院 妇产科胎儿中心,广东 广州 510080)
胎儿单基因遗传病的无创检测技术研究进展
周祎 蒋馥蔓 王子莲
(中山大学附属第一医院 妇产科胎儿中心,广东 广州 510080)
1 单基因病和NIPT技术(non-invasive prenatal test)
单基因疾病(monogenedisease)是指由一对等位基因控制的疾病或临床病理表现,即按照孟德尔方式传递的疾病,也称孟德尔遗传病(medeliandisease)。根据致病基因所在染色体的不同可将单基因病分为常染色体遗传病、性染色体遗传病和线粒体遗传病,性染色体遗传病包括X染色体遗传病和Y染色体遗传病。根据致病基因表现型的不同可将单基因分为显性遗传病和隐性遗传病。截止到2017年1月20日,OMIM(online medelian inheritance in man)数据库已收入单基因病8000多种,遗传发病机制明确的单基因病近5000种。根据世界卫生组织(WHO)统计,全球出生人口所有单基因病的发病率约为10/1000,常见的单基因病有地中海贫血、镰状细胞贫血、血友病、囊胞性纤维症、脆性X综合征、亨廷顿舞蹈病、软骨发育不全、苯丙酮尿症、G6PD缺乏症等。单基因病大多没有治愈方法,并且治疗成本高,因此进行产前遗传检测是预防患单基因病以及提前进行临床干预的主要方法。
检测胎儿单基因遗传病最为直接的方法是获取胎儿组织或细胞进行检测。临床上常用的方法是通过绒毛膜取样和羊膜穿刺获取胎儿细胞进行产前诊断,但这两种方式均为有创取样,会伴随一定的流产风险[1]。此外,羊膜穿刺一般在妊娠中期进行,可能会拖延后续的临床干预[2]。孕妇外周血中,胎儿游离DNA(cell free fetal DNA, cfDNA)的发现[3],使胎儿遗传病无创检测有了理论可能性。NIPT是基于胎儿游离DNA进行胎儿遗传病无创检测的成功应用,NIPT检测范围是胎儿染色体异常情况,检测的分辨率处在染色体水平和较大片段的缺失重复,而对于点突变或者小片段的缺失重复等单基因病的致病变异无法进行准确的检测[5,6]。
利用胎儿游离DNA进行单基因病的检测技术发展主要受制于3个因素。首先,根据单基因病的遗传特点,不同类型的单基因病致病原因的复杂程度不同,因而检测的策略各有差异;第二,母体外周血中胎儿游离DNA含量很低(10%左右)[4,7],母亲自身的游离DNA给检测带来很大的背景噪声,从而使得母源性变异无法进行检测;第三,低浓度的胎儿游离DNA会导致无法检出结果或造成假阴性与假阳性结果等。随着检测技术的发展,近年来单基因病无创检测技术有了飞快的发展,即胎儿染色体非整倍性无创检测技术的成功应用之后,将成为研究和临床领域下一个技术突破点。
2 非母亲来源的变异检测
在母体外周血DNA中,找到与母亲自身携带的遗传信息不同的序列或变异信息,可以直接或间接地检测出胎儿的遗传变异情况。父源特异性的非致病序列可以作为遗传检测的标记物,可用于排除某些性别连锁的单基因遗传病。父源特异性的致病序列以及胎儿新发遗传变异的检测可以直接用于胎儿单基因病遗传分析。
2.1 父源变异检测-性染色体检测策略 胎儿cfDNA的发现是在母体血浆中发现了胎儿来源的Y染色体片段[3]。因此父源特异性变异检测是cfDNA用于胎儿遗传病检测的早期探索应用。检测Y染色体特有的基因或者遗传序列,一方面用于性别判定,一方面用于检测性染色体连锁的单基因遗传病。采用PCR技术通过检测母亲外周血中的Y染色体特异基因SRY或DSY14获取胎儿性别[10]。如果胎儿是女胎则可以排除诸如血友病的X染色体连锁隐性遗传病。而在NIPT中,定量检测Y染色体片段含量,除了性别判断之外,还可以估算出母体血浆中胎儿游离DNA的浓度[11],而这一指标是影响单基因病无创检测技术发展的一个重要因素。此外,有研究利用X染色体上的父源特异性短串联重复序列作为遗传标记物来获取女胎遗传信息[12]。
2.2 父源变异检测-突变基因连锁SNP检测策略 除了上述针对性染色体遗传信息进行检测的策略外,对于常染色体而言,寻找与母亲遗传信息不同的父源特异性变异也是最直接、最早的研究的思路之一。显性单基因病中,若发现父源变异信息,则可以判断为患病胎儿。隐性单基因病中,若未发现父源特异性变异信息,则可以排除胎儿患病或者患儿为重症患儿的情况[13]。另一方面,若父亲和母亲携带相同变异,还可以通过寻找与父源变异连锁的SNP来检测胎儿是否携带父源变异,进而对胎儿患病情况进行判定[15]。Ding等[14]利用与HBB突变连锁的父源特异性SNP位点检测胎儿游离DNA中父亲来源的变异。父母双方的SNP位点信息充足的情况下,父亲来源的HBB突变与rs2187610snp位点的连锁关系被确定,采用SABER方法可以检测出SNP位点连锁的HBB突变,进而确定胎儿的单倍体型,该方法可以排除重型β-地中海贫血突变。特点是无论父母双方是否携带同一种HBB突变,都可以通过与HBB连锁的SNP位点来排除父源性的突变,进而排除胎儿患重型β-地中海贫血突变的可能。局限在于需要更多地发现与HBB突变连锁的特异性SNP位点来保证检测的适用性和准确度。
全基因组深度测序对胎儿单基因遗传病进行检测的方法拓展了这个策略的应用,Lo等[15]选择父亲杂合母亲纯合的SNP位点进行母体血浆中父亲来源的等位基因检测。如果母体外周中检测到父亲特异性的等位基因可以确定胎儿遗传了父源特异性基因,如果未检测到则可以推断胎儿遗传了父母同型等位基因。该方法的特点是SNP位点在全基因组范围内分布,并且通过一种二项分布建模(binomial mixture model)提高了检测的准确率。
2.3 胎儿新发变异检测胎儿新发变异的检测策略 与父源特异性变异检测的思路一致,都是检测母亲自身不存在的变异信息。传统的PCR技术以设计针对特定变异类型的探针为基础,并不能准确检测出胎儿新发变异。高通量测序技术使得获取胎儿全基因组信息成为可能,但是由于新发变异率很低以及高通量测序中存在的错误率,胎儿新发变异的检出率和检测效果并不理想[16]。Lo等[15]通过提高全基因组的测序深度结合多步过滤假阳性突变的生物信息学分析方法提高了胎儿新发变异检测准确度和敏感度。
检测非母源性变异和特异序列的策略,通过简单的PCR技术即可进行检测,其应用范围在本质上属于定性检测的范畴,并且检测建立在已知特异性标记序列或变异的基础上。进行单基因病无创检测受到胎儿cfDNA浓度的影响,而无论是父源特异性变异还是胎儿特异性变异的定量,均和胎儿游离DNA含量直接相关,因此可能会因为胎儿浓度过低,而导致检测错误[4,6]。另外,利用父源特异SNP进行检测的策略中,需要提前获取与父源特异突变连锁的SNP位点,并且需要足够多的SNP位点信息来排除PCR和测序过程中出现的错误,以及因胎儿cfDNA含量过低导致的检测失败[4,13,14]。
3 母亲来源的变异检测
检测胎儿游离DNA是否母亲来源的变异难度很大。首先,区分母亲自身的DNA与胎儿游离DNA中母亲来源的DNA,尚无特定的分子标记进行区分;其次,孕妇外周血中,母亲自身的DNA片段高达90%,而胎儿游离DNA占比仅为10%左右[5],无法采用检测特异性突变的方式获得准确的结果。基于NIPT的理论基础,主要采用定量检测策略,通过构建胎儿的单倍体型,进而判定胎儿是否携带了母亲来源的变异。目前主要的策略有3种:RMD策略[17]、RHDO策略[18,19]和GRAD策略[15]。
3.1 RMD策略 RMD策略是相对突变剂量(relative mutation dosage)检测方法的简称。该方法是在用于NIPT检测中的RCD法[18]的基础上的延伸,其原理见图1所示。通过计算突变等位基因和野生型等位基因的比率进而计算出母亲和胎儿对外周血游离DNA的贡献。比如,母体外周血含有100个基因组当量(GE),其中胎儿游离DNA占10%,那么母亲来源的游离DNA为90GE,假设母亲的基因型为MN,如果胎儿基因型为NN,那么,母亲本身对等位基因M和N的贡献均为90个拷贝,而胎儿对等位基因N贡献为20,进而可以计算出M/N=9∶11。通过比较等位基因M和N的相对剂量可以获得胎儿的单倍体型信息。Lun等[15]在2008年首次介绍了RMD策略结合序贯概率比(SPRT)计算的方法,成功检测出β-地中海贫血HBB基因两种突变的信息。该研究中利用的是dPCR技术,除了RMD策略,还开发了一种cfDNA富集方法——核酸片段选择,用于解决胎儿游离DNA浓度过低问题。该方法受限于dPCR的高成本和设计特异性探针的复杂性,在临床推广应用中具有一定的局限性;但是该方法的策略思路及其在胎儿游离DNA片段富集的成功探索为后续的研究提供了基础。
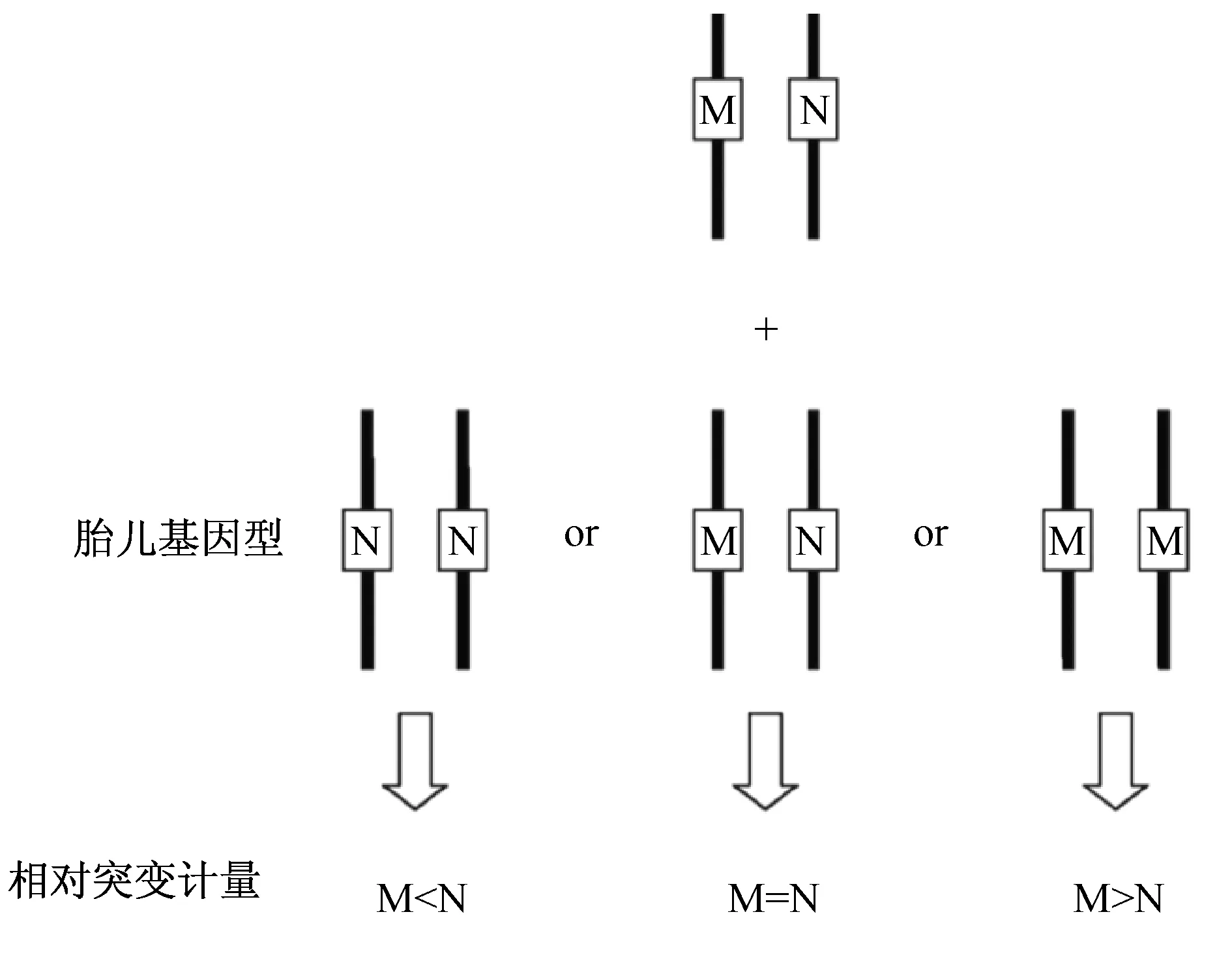
图1 RMD策略原理示意图[17]
3.2 RHDO策略 RHDO(relative haplotype dosage) 是对胎儿所遗传的父母双方的单倍体型剂量进行相对定量分析的方法[2,16,17]。其原理如图2所示。选取父母双亲的SNP位点用来构建两组不同的单倍体型(HapⅠ和HapⅡ),其中父亲的基因型为纯合,母亲的基因型为杂合。胎儿所遗传的单倍体型,一半来自父亲,一半来自母亲。在母亲外周血中,母亲自身的单倍体型为杂合,而胎儿的单倍体型则有杂合和纯合两种情况。胎儿单倍体型为纯合时,母体血浆中HapⅠ和HapⅡ的剂量会出现不平衡。那么通过 SPRT(sequential probability ratio test)序贯概率比检验计算母体外周血中HapⅠ和HapⅡ的相对含量,则可以推断出胎儿的遗传信息。
Lo等[16]利用高通量测序对母体外周血游离DNA进行检测,证明了在母体血浆中检测胎儿全基因组以及检测胎儿单基因遗传病的可行性。该研究选取了携带两种不同类型β-地中海贫血突变的夫妇,检测其胎儿的遗传情况。其中父亲来源的变异利用父源特异性SNP进行检测,而母亲来源的变异则采用RHDO策略进行检测。采用这种胎儿全基因组检测的方法进行单基因病的无创检测的前提是已知父母的变异类型以及特异性的SNP位点。在这项概念验证研究中,母亲的单倍体型是通过胎儿绒毛膜取样获得胎儿遗传物质而获取的,在实际临床应用中则需要通过家族先证者的遗传信息或者来自数据库中的已知信息来解决这一问题。只利用了基因组区域中父亲纯合而母亲杂合的等位基因SNP位点,对由于始祖效应或近亲婚姻导致的父母双方致病基因区域单倍体型结构非常相似的情况却没有提及。并且检测的分辨率还受到测序深度和胎儿游离DNA浓度的影响,全基因组测序的成本也是限制该方法临床应用的瓶颈。但是基于全基因组测序RHDO策略相对于前述基于dPCR技术的RMD策略具有一定的优势。一方面,全基因组范围的检测使的该技术能够拓展应用到多种类型的单基因病检测中。另一方面,利用多个变异构成的单倍体型进行检测相对于仅检测一种变异,可以获得更多的遗传信息,使的在较低深度下可以获得检测结果。
Lam等[17]进一步优化了基于高通量测序技术的RHDO策略。该研究先利用dPCR技术获得了父母双亲的单倍体型信息,进而对目标区域进行靶向靶向测序,解决了全基因组测序的高成本限制。对父母双方单倍体型基因上的SNP位点进行分类(type α和type β),再综合判断胎儿是否携带了双亲的致病突变位点,解决了父母双方SNP位点均为杂合时的问题。较之前的检测分析方法相比,本研究采用的实验方法和分析流程更加适宜用于临床检测,而且成本价格效益更高。
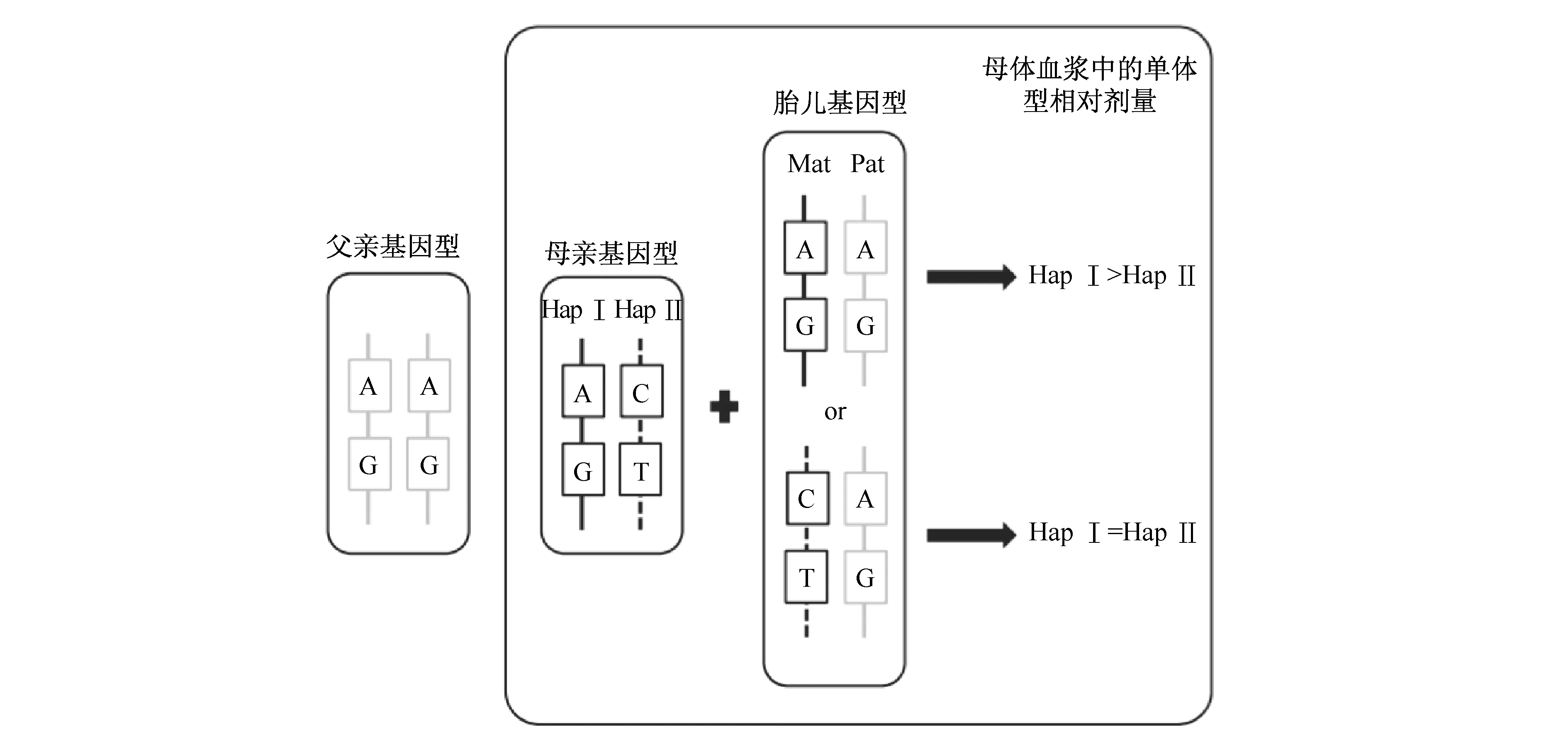
图2 RHDO策略原理示意图[4]
3.3 深度测序GRAD策略 GRAD(genome-wide relative allelic dosage) 全基因组相对等位基因剂量是RMD策略思想与全基因组测序技术结合的方法[13]。GRAD策略可以通过深度测序获得的高质量基因组信息直接检测母体血浆中杂合等位基因的序列信息,进而通过SPRT方法检测出母体中杂合等位基因的相对含量,从而在原理上与RMD策略相同。而与前述全基因组测序RHDO策略不同的是GRAD无需构建母亲的单倍体型,并且高深度全基因组测序将检测的分辨率提升了约90倍。RHDO全基因组测序中检测出母亲来源的单倍体型区域为7332[16],而GRAD全基因组测序中检测出母亲来源的杂合SNP为656676[13]。利用该方法成功检测出心面综合征胎儿cfDNA中母源性遗传信息,准确率达到96.8%。此外,该研究对母体血浆采用无PCR扩增的建库方法,证实母体血浆的游离DNA片段末端具有一定的规律性特点。某些片段末端倾向性的与胎儿游离DNA或母亲游离DNA片段相关,利用这个特点可以进行胎儿DNA浓度的估计。GRAD策略应用受到测序深度、特异性SNP数量以及胎儿游离DNA浓度的影响。尤其是高深度测序的高成本仍然是其临床应用的一大限制,但随着测序成本的降低,这个策略具有明显的应用优势。另外,无PCR扩增的建库方法的成功应用,也为三代测序技术(单分子实时测序)在无创单基因病检测中的应用开启了研究方向。
4 总结和展望
母体外周血中游离DNA的发现使无创产前检测技术有了实质性进展,而高通量分子检测技术,尤其是NGS技术的发展使NIPT在研究和临床检测中实现了广泛应用。NIPT技术与胎儿cfDNA结合使无创单基因病检测具有了理论上的可能性。不同于胎儿染色体非整倍性的无创检测,胎儿单基因病无创检测对检测技术、检测策略的要求更高,技术难点也更多。在技术上,依靠胎儿游离DNA进行单基因病检测的发展经历了从普通PCR、qPCR、dPCR、到低深度NGS,再到高深度NGS的过程,未来三代测序技术(单分子实时测序技术)的发展应用将成为后续研究的一个方向。而就目前的研究进展来看NGS技术以其通量大、应用灵活、可拓展性好、成本不断降低等特点,在单基因病无创检测的临床应用中具备一定的优势。在检测策略上,从定性排除发展到了定量检测。尤其是对母亲来源DNA的检测从无法检测,到通过构建单倍体型检测,发展到了不依赖单倍体型的检测方法。检测范围从单个突变位点和单个序列扩展到了全基因组的范围。因此,胎儿单基因病的无创检测技术将成为未来研究的重点和有望突破的热点。
[1] Mujezinovic F, Alfirevic Z.Procedure-related complications of amniocentesis and chorionic villous sampling:a systematic review[J].Obstet Gynecol, 2007, 110(3):687-694.
[2] Han DSC, Lo YMD.Non‐invasive prenatal testing of monogenic fetal characteristics by maternal plasma DNA analysis[J].ISBT Science Series, 2015, 10(S1):197-205.
[3] Lo YMD, Corbetta N, Chamberlain PF, et al.Presence of fetal DNA in maternal plasma and serum[J].Lancet, 1997, 350:485 -487.
[4] Chiu RWK, Cantor CR, Lo YMD.Non-invasive prenatal diagnosis by single molecule counting technologies[J].Trends in genetics, 2009, 25(7):324-331.
[5] Chiu RWK, Chan KCA, Gao Y, et al.Noninvasive prenatal diagnosis of fetal chromosomal aneuploidy by massively parallel genomic sequencing of DNA in maternal plasma[J].Proceedings of the National Academy of Sciences, 2008, 105(51):20458-20463.
[6] Peters D, Chu T, Yatsenko SA, et al.Noninvasive prenatal diagnosis of a fetal microdeletion syndrome[J].New England Journal of Medicine, 2011, 365(19):1847-1848.
[7] Lo YM, Tein MS, Lau TK.et al.Quantitative analysis of fetal DNA in maternal plasma and serum:implications for noninvasive prenatal diagnosis[J].Am J Hum Genet, 1998, 62:768-775.
[8] Wright CF, Wei Y, Higgins JPT, et al.Non-invasive prenatal diagnostic test accuracy for fetal sex using cell-free DNA a review and meta-analysis[J].BMC Research Notes, 2012, 5(1):476.
[9] Jiang F, Ren J, Chen F, et al.Noninvasive Fetal Trisomy (NIFTY) test:an advanced noninvasive prenatal diagnosis methodology for fetal autosomal and sex chromosomal aneuploidies[J].BMC Medical Genomics, 2012, 5(1):57.
[10] Tang NLS, Leung TN, Zhang J, et al.Detection of fetal- derived paternally inherited X-chromosome polymorphisms in maternal plasma[J].Clin Chem,1999,45:2033-2035.
[11] Chiu RWK, Lau TK, Leung TN, et al.Prenatal exclusion of b- thalassaemia major by examintion of maternal plasma[J].Lancet, 2002, 360:998-1000.
[12] Ding C, Chiu RWK, Lau TK, et al.MS analysis of single-nucleotide differences in circulating nucleic acids:application to noninvasive prenatal diagnosis[J].Proc Natl AcadSci U S A, 2004, 101(29):10762-10767.
[13] Chan KCA, Jiang P, Sun K, et al.Second generation noninvasive fetal genome analysis reveals de novo mutations, single-base parental inheritance, and preferred DNA ends[J].Proc Natl AcadSci U S A, 2016, 113(50):E8159-E8168.
[14] Kitzman JO, Snyder MW, Ventura M, et al.Noninvasive whole-genome sequencing of a human fetus[J].Sci Transl Med,2012 4(137):137ra76.
[15] Lun FMF, Tsui NBY, Chan KCA, et al.Noninvasive prenatal diagnosis of monogenic diseases by digital size selection and relative mutation dosage on DNA in maternal plasma[J].Proceedings of the National Academy of Sciences, 2008, 105(50):19920-19925.
[16] Lo YMD, Chan KCA, Sun H, et al.Maternal plasma DNA sequencing reveals the genome-wide genetic and mutational profile of the fetus[J].Science Translational Medicine, 2010, 2(61):61ra91-61ra91.
[17] Lam KWG, Jiang P, Liao GJW, et al.Noninvasive prenatal diagnosis of monogenic diseases by targeted massively parallel sequencing of maternal plasma:application to β-thalassemia[J].Clinical chemistry, 2012, 58(10):1467-1475.
[18] Lo YMD, et al.(2007) Digital PCR for the molecular detection of fetal chromosomal aneuploidy.Proc Natl Acad Sci USA 104:13116-13121.
R714.53
A
2017-04-30)
编辑:宋文颖
10.13470/j.cnki.cjpd.2017.02.001
猜你喜欢
实用手外科杂志(2022年2期)2022-08-31
科学导报·学术(2020年29期)2020-10-21
趣味(数学)(2020年4期)2020-07-27
支部建设(2020年15期)2020-07-08
美与时代·美术学刊(2019年9期)2019-11-29
中学生理科应试(2019年4期)2019-07-08
中国生殖健康(2019年3期)2019-02-01
百科知识(2015年18期)2015-09-10
外语教学理论与实践(2014年2期)2014-06-21
中学生物学(2008年12期)2008-12-27
