
超高效液相色谱-串联质谱法快速测定沉积物中11种藻毒素
2017-09-26 06:16:39张付海田丙正
分析测试学报 2017年9期
赵 彬,王 昭,张 敏*,张付海,田丙正,丁 磊
(1.安徽省环境监测中心站,安徽 合肥 230071;2.安徽省出入境检验检疫局 检验检疫技术中心,安徽 合肥 230031)
超高效液相色谱-串联质谱法快速测定沉积物中11种藻毒素
赵 彬1,王 昭2,张 敏1*,张付海1,田丙正1,丁 磊2
(1.安徽省环境监测中心站,安徽 合肥 230071;2.安徽省出入境检验检疫局 检验检疫技术中心,安徽 合肥 230031)
建立了超高效液相色谱-串联质谱(UPLC-MS/MS)快速测定沉积物中11种藻毒素的方法。沉积物经冷冻干燥、粉碎过筛,用0.1 mol/L EDTA-Na4P2O7溶液涡旋超声提取,经HLB固相萃取小柱净化后,用甲醇-0.2%甲酸洗脱、浓缩并氮吹定容至1 mL。经Waters BEH C18色谱小柱,以乙腈-0.2%甲酸水溶液为流动相,梯度洗脱分离后,在电喷雾正离子模式下,以超高效液相色谱-串联质谱多级监测模式(MRM)外标法进行定性定量分析。结果表明:沉积物中11种藻毒素的检出限为1.0~5.0 ng/kg。对同一环境样品进行了0.1、1.0、4.0 μg/kg不同水平的加标回收试验,平均回收率为70.3%~112.5%,相对标准偏差(RSD)为2.2%~9.3%。该方法快速、灵敏、准确,可应用于沉积物中11种藻毒素的快速监测。
沉积物;藻毒素;超高效液相色谱-串联质谱
藻毒素是蓝藻产生的毒性很强的天然毒素。藻毒素可分为两大类:一类具有急性致死作用,主要包括神经毒素(Neurotoxins)和肝毒素(Hepatotoxins);另一类对动物无较强致死毒性,但具有较高的特异生物活性,如细胞毒素(Cytotoxins)。神经毒素和肝毒素的危害最大,文献中对二者的报道最多[1]。微囊藻毒素(Microcystins,MCs)、节球藻毒素(Nodularins,NOD)、拟柱胞藻毒素 (Cylindrospermospins,CYN)是分布最为广泛的3种毒素。其中微囊藻毒素和节球藻毒素属于肝毒素,其主要靶器官为肝脏。微囊藻毒素是由微囊藻产生的对蛋白磷酸酶PP1和PP2A具有抑制性的环状七肽,目前已知的种类有90多种。节球藻毒素主要由泡沫节球藻(Nodulariaspumigena)产生,其结构与微囊藻毒素类似,是一种环状五肽结构,可抑制蛋白磷酸酶PP1和PP2A的活性。拟柱胞藻毒素的分子式为C15H21N5O7S,分子量为415.4,易溶于水、甲醇、二甲亚砜,是具有细胞毒性、肝毒性、神经毒性和遗传毒性的生物碱毒素,可通过抑制蛋白质合成而导致肠胃炎、肝损伤、肾损伤、肠损伤,危及人体健康。近年来随着人们生活水平的提高和工农业活动的发展,水体富营养化现象日趋普遍,世界上淡水湖泊蓝藻爆发的频率也不断增加[2-4]。
巢湖是我国第五大淡水湖,每年蓝藻不时爆发,一直备受关注。在实际治理过程中,蓝藻在水华时常被拦截、打捞作为有机肥施入农田,或堆放在湖岸边沉降到湖底。而藻细胞破裂释放高浓度的藻毒素会严重污染沉积物,会危害相关水生生物的生长和相关水产品的安全,进而通过食物链对人体健康产生危害[5]。与水体环境相比,沉积物由于基质更加复杂,样品采集及处理过程更加繁琐,其藻毒素的监测方法鲜有研究和报道[6]。因此,建立快速、准确、灵敏可靠的沉积物中藻毒素的测定方法显得尤为重要。
目前该类化合物的测定只针对微囊藻毒素类、节球藻毒素或者拟柱胞藻毒素分别进行,其检测方法主要有生物化学法、酶联免疫检测法、高效液相色谱法和液相色谱质谱联用法等[7-13],尚未见使用一种方法对该3种毒素进行同时检测的报道。生物化学法选择性较差,灵敏度不够,只能测定毒素总量,无法分别测定不同种类毒素含量。酶联免疫法假阳性几率大,且重现性差。液相色谱法则由于环境水样基质复杂,背景干扰多等特点,不易定性,且易出现假阳性。而液相色谱-串联质谱法则能更好地应对基质复杂,有背景干扰的样品监测要求。针对基质复杂、含水率较高的环境样品的前处理方法主要有液-液萃取、加速溶剂萃取(ASE)、固相小柱净化(SPE)和渗透凝胶色谱净化(GPC)等。本文在前期研究的基础上[14-15],建立了同步提取、同时测定沉积物中11种藻毒素的超高效液相色谱-串联质谱快速检测方法。
1 实验部分
1.1 仪器与试剂
真空冷冻干燥机(LGJ-25,北京四环科学仪器);台式鄂式破碎仪(JC6,北京格瑞德曼);Waters TQ-S超高效液相色谱-串联质谱(Waters公司,美国);Masslynx 4.1工作站;固相萃取柱C18、HLB(6 mL/500 mg,Waters公司);定量平行浓缩仪(MultiVap-8,北京莱伯泰科);超纯水制备仪(Millipore Milli-Q Integral15,美国);色谱柱:Waters BEH C18(50 mm×2.1 mm,i.d.,1.7 μm)。
11种藻毒素标准品购于瑞士ENZO@Life Sciences公司,100 μg,纯度≥95.0%(化合物信息详见表1),甲酸、甲醇、乙腈(农残级,Merk公司);超纯水;0.22 μm有机滤头。
1.2 样品的采集与制备
采集2 kg沉积物于棕色光口玻璃瓶中,4 ℃冷藏保存。样品经真空冷冻干燥后,研磨粉碎过0.24 mm孔径筛。
1.3 样品前处理
1.3.1样品提取准确称取10.00 g上述制备好的样品于100 mL离心管中,分3次加入100 mL 0.1 mol/L EDTA-Na4P2O7溶液,涡旋混匀后超声萃取20 min。然后以8 000 r/min离心10 min,合并收集提取液,待净化。
1.3.2固相萃取小柱净化用5 mL甲醇、10 mL水活化HLB固相萃取柱,保证小柱柱头浸润。将上述提取液以小于5 mL/min 的流速通过小柱富集,再用10 mL水淋洗,将小柱上保留较弱的杂质淋洗下来。用氮气吹扫小柱5 min,将小柱中的残留水分完全去除。再用15 mL甲醇-0.2%甲酸水溶液(体积比9∶1)洗脱富集后的小柱,洗脱液收集于接收管中。将上述洗脱液氮吹浓缩至近干,加入甲醇-0.2%甲酸水溶液(1∶4)定容至1.0 mL,混匀后过0.22 μm滤膜,待测。
1.4 色谱质谱条件
色谱条件:流动相:A为乙腈,B为0.2%甲酸-水。采用线性梯度洗脱条件:0~0.5 min,75%B;0.5~4.5 min,75%~45%B; 4.5~5.0 min,45%~5%B;5.0~6.0 min,5%B;6.0~6.5 min,5%~75%B;6.5~7.0 min,75%B。进样量为10 μL。流速:0.4 mL/min。样品室温度:10 ℃;色谱柱温:40 ℃。
质谱条件:采用电喷雾离子源(ESI)正离子模式进行检测,多级反应监测模式(MRM)。喷射电压1.5 kV,源温度为150 ℃,脱溶剂气温度为450 ℃,流量为800 L/h,锥孔气流速为150 L/h,锥孔电压和碰撞能量见表1。

表1 11种藻毒素的名称、CAS号、分子量和MRM分析条件Table 1 The names,CAS numbers,molecular weight and MRM analysis conditions of 11 kinds of algal toxins
*quantitative ion
2 结果与讨论
2.1 样品前处理条件的优化
2.1.1样品前处理方法的选择目前关于环境沉积物样品的前处理大都采用风干干燥的方法,但该法由于需大量时间而可能导致目标物的降解损失。因此本实验选择真空冷冻干燥法,可以快速对含水率较高的沉积物进行干燥。
对于干燥研磨好的样品,目前的提取方法大多为液-液萃取、超声萃取和加速溶剂萃取(ASE)等。本文参考文献方法[6],超声提取20 min,能够更加快速并充分提取目标化合物,11种藻毒素的提取回收率为68.6%~109.1%,相比较文献报道[6]的54.9%~97.4%,回收率提高了12%~25%。
环境样品基质复杂,背景干扰物多,目前常用的净化方法有固相小柱萃取法(SPE)、凝胶渗透色谱法(GPC)、基质解析法等。由于这11种藻毒素为极易溶于水、难溶于有机溶剂的多肽类化合物,故不宜选用以有机溶剂为流动相的GPC净化方法。固相小柱萃取(SPE)是一种常用的样品前处理净化技术,是液固萃取和液相色谱柱技术相结合的产物,能很好地实现目标物与样品基体和干扰物的分离。本实验对比了常用SPE小柱C18和HLB柱的净化效果(图1),发现HLB柱的净化效果及回收率优于C18柱。其中拟柱胞藻毒素(CYN)由于极易溶于水,分子量小于其它几种藻毒素,而很难在固相萃取柱上得到有效保留,所以回收率不高,但可以通过直接进样或重复萃取富集步骤的方法提高其回收率(重复萃取富集步骤3次即可获得较好的回收率)。综上所述,本实验最终选取HLB柱进行样品净化富集。

图1 不同净化方法效果的比较Fig.1 Effect comparison of different purification methods

图2 两种不同液相色谱小柱的色谱图分离比较Fig.2 Chromatograms separation on two different liquid chromatographic columns
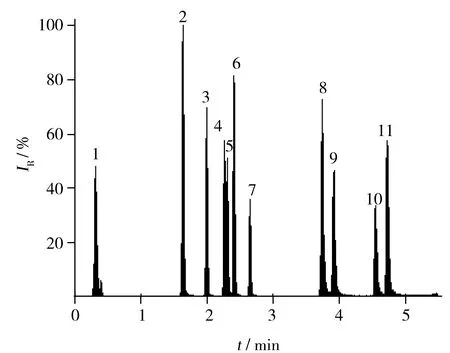
图3 11种藻毒素MRM的TIC色谱图Fig.3 MRM TIC chromatogram of 11 kinds of algal toxins1.CYN,2.MC-RR,3.NOD,4.MC-YR,5.MC-HtyR,6.MC-LR,7.MC-WR,8.MC-LA,9.MC-LY,10.MC-LW,11.MC-LF
2.1.2洗脱液及其用量的优化对于极性样品,常采用甲醇、乙腈等亲水性有机溶剂作为洗脱液。因此本文优化了不同洗脱液及其用量。结果发现,15 mL甲醇-0.2%甲酸水溶液(9∶1)的洗脱效果最佳。在甲醇中添加不同含量的甲酸(0.05%、0.1%、0.2%、0.3%、0.4%)进行洗脱,发现甲酸含量在0.05%~0.2%范围时的回收率逐渐增加。这可能是因为加入甲酸后,藻毒素在酸性环境下多肽质子化,同时也减少了其与硅胶表面硅醇基之间的相互作用,使其更易被洗脱,所以最终选择甲醇-0.2%甲酸水溶液为洗脱液。
2.2 色谱及质谱条件的优化
2.2.1色谱条件的优化分别比较了不同类型的反相色谱柱(Waters BEH C18、Waters HSS T3、Waters HSS C8)对待测物质的色谱分离效果。结果表明(图2),拟柱胞藻毒素在Waters BEH C18色谱柱上几乎不保留,在Waters HSS T3色谱柱上有保留,但其它藻毒素不能达到较好的分离。而其它10种藻毒素在Waters BEH C18色谱柱(50 mm×2.1 mm,i.d.1.7 μm)上获得了理想的分离效果。综合考虑,选用Waters BEH C18色谱柱作为分析柱。
甲醇和乙腈是液相色谱常用的流动相。在优化的色谱分离条件下,考察了甲醇-水和乙腈-水流动相体系对目标物离子化程度的影响。实验发现乙腈-水的仪器响应值高于甲醇-水。由于目标物的基本结构是7个氨基酸组成的单环多肽,在正离子电离模式下溶液中能够电离出H+。所以,在流动相中适当加入酸性物质可以促进目标物的电离,从而获得较高的离子化效率。因此本文选取0.2%甲酸-水作为流动相梯度洗脱,并在5 min内实现了目标化合物的快速分离,获得了最佳实验结果。
2.2.2质谱条件的优化根据微囊藻毒素具有多肽质子化的化学电离性质,本文选用正电离模式(ESI+),采用蠕动泵直接进样的方式进行质谱条件优化。根据CAC和EU第657/2002/EEC号决议有关规定,选择两对离子监测MRM即可满足要求。通过仪器自带软件Masslynx 4.1的自动优化软件IntelliStart,对目标物的锥孔电压、碰撞能、定量离子和辅助定性离子进行了优化。11种藻毒素的TIC色谱图见图3。
2.3 标准曲线、线性范围及检出限
将标准储备液用流动相梯度稀释成6种不同质量浓度的标准溶液(0.5、1、5、10、20、50 μg/L),采用本方法测定,以标准物的质量浓度(x,μg/L)为横坐标,对应峰面积(y)为纵坐标进行相关性回归分析,得回归方程及相关系数。并根据HJ168-2010中方法检出限的一般确定方法,依据公式MDL=t(n-1,0.99)×S,通过重复空白试验7次计算得各组分的检出限为1.0~5.0 ng/kg,结果见表2。
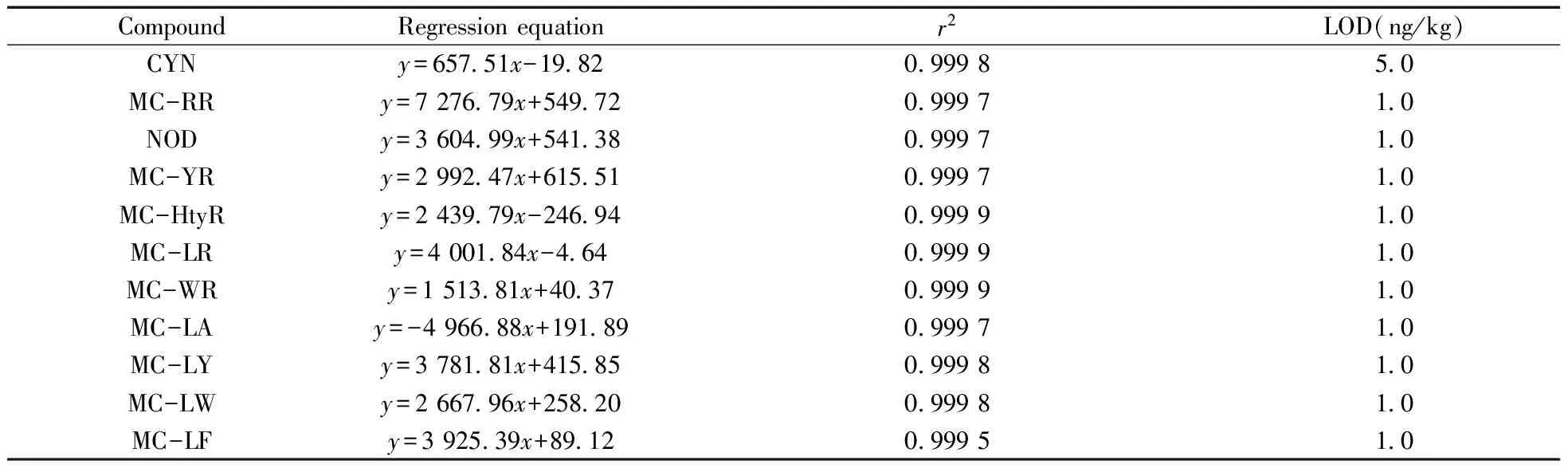
表2 11种藻毒素的标准方程、相关系数、仪器检出限及方法检出限Table 2 Regression equation,correlation coefficient,instrument detection limit and method detection limit of 11 kinds of algal toxins
2.4 方法的回收率与精密度
对一未检出目标物的实际环境样品进行加标回收率和精密度实验,样品分别添加0.1、1.0、4.0 μg/kg 3个水平的标准溶液,每个加标水平平行6次,按本方法进行测定。计算得方法回收率及相对标准偏差(RSD)结果见表3。

表3 11种藻毒素的加标回收率和相对标准偏差(n=6)Table 3 Recoveries and relative standard deviations of 11 kinds of algal toxins(n=6)
2.5 样品的测定

图4 实际样品的TIC色谱图Fig.4 TIC chromatogram of real sample
于12个不同时期在环巢湖湖面及入湖河流的23个监测点位采集了沉积物,应用本方法进行分析测定,实际样品谱图见图4。分析结果表明,有14组样品检出4种藻毒素(MC-LR,MC-RR,MC-LW和MC-LF),含量为19.3~127.4 ng/kg。
3 结 论
建立了超高效液相色谱串联质谱联用技术同时快速测定沉积物中11种藻毒素的方法。沉积物经冷冻干燥、粉碎过筛,超声提取,经HLB固相萃取小柱净化,超高效液相色谱分离,串联质谱电喷雾正离子模式下采用多级离子监测方式检测,外标法定量。该方法检出限为1.0~5.0 ng/kg,优于已有文献[6]。对巢湖湖区及入湖河流的沉积物进行了研究调查,结果显示,在不同时期均不同程度地检出了相关藻毒素。该方法快速、灵敏、准确,可有效应用于沉积物中藻毒素的实际监测。
[1] Duy T N,Lam P K S,Shaw G R,Connell D W.Rev.Environ.Contam.Toxicol.,2000,163:113-186.
[2] Pérez S,Aga D S.J.TrendsAnal.Chem.2005,24(7):658-670.
[3] Ward C J,Beattie K A,Lee Y C,Codd G A.FEMSMicrobiologyLetters,1997,152: 465-473.
[4] McElhiney J,Lawton L A.ToxicologyandAppliedPharmacology,2005,203: 219-230.
[5] Zhan X J,Xiang L,Li Y W.Chin.Environ.Sci.(詹晓静,向垒,李彦文.中国环境科学),2015,35(7) : 2129-2136.
[6] Li Y W,Huang X P,Wu X L.Chin.J.Anal.Chem.(李彦文,黄献培,吴小莲.分析化学),2013,41(1): 88-92.
[7] Jiang M,Xu H.ActaEcol.Sinica(江敏,许慧.生态学报),2014,34(16) : 4473-4479.
[8] Wu W W,Yang Z J,Gu H F.J.Instrum.Anal.(吴伟文,杨左军,顾浩飞.分析测试学报),2007,26(4):545-547.
[9] Guo J,Yang X L,Ye M L.Chin.J.Anal.Chem.(郭坚,杨新磊,叶明立.分析化学),2011,39(8):1256-1260.
[10] Li Z X,Zhao Q N,Zhang C L.Chin.J.Anal.Lab.(黎志轩,赵倩宁,张长立.分析试验室),2012,11(31):90-93.
[11] Yu R P,Tao G J,Qin F.Chin.J.Anal.Chem.(虞锐鹏,陶冠军,秦方.分析化学),2003,31(12):1462-1464.
[12] Zhang M,Tang F L,Chen F.Chin.J.Chromatogr.(张明,唐访良,陈锋.色谱),2012,30(1):51-55.
[13] Zhang C Y,Zhao X R,Zheng X Z.Environ.Chem.(张春燕,赵兴茹,郑学忠.环境化学),2012,31(10): 1663-1664.
[14] Zhao B,Zhang M,Zhang F H,Hu Y Q,Tian B Z,Wang X.Chem.Anal.Meter.(赵彬,张敏,张付海,胡雅琴,田丙正,王鑫.化学分析计量),2016,25(2): 48-51.
[15] Zhao B,Zhang M,Zhang F H.Meteo.Environ.Res.,2016,7(6):54-57.
Rapid Determination of 11 Algal Toxins in Sediments by Ultra Performance Liquid Chromatography-Tandem Mass Spectrometry
ZHAO Bin1,WANG Zhao2,ZHANG Min1*,ZHANG Fu-hai1,TIAN Bing-zheng1,DING Lei2
(1.Anhui Environmental Monitoring Center,Hefei 230071,China;2.Inspection and Quarantine Technology Center,Anhui Entry-Exit Inspection and Quarantine Bureau,Hefei 230031,China)
A method was established for the rapid determination of 11 algal toxins in sediments by ultra performance liquid chromatography - tandem mass spectrometry.Firstly,the sample was freezingly dried out,and then sieved,followed by purification with 0.1 mol/L EDTA-Na4P2O7solution and HLB solid phase extraction column,elution with methanol-0.2% formic acid,and finally concentrated and mixed with nitrogen to 1 mL.The separation of the analyte was performed on a Waters BEH C18chromatographic column with acetonitrile -0.2% formic acid solution as mobile phase by gradient elution.The sample was analyzed by ultra performance liquid chromatography-tandem mass spectrometry in positive ions electrospray mode under multi-stage monitoring mode (MRM),and quantified by external standard method.The results showed that the detection limits for 11 algal toxins in sediment were in the range of 1.0-5.0 ng/kg.The average recoveries for the same environmental samples at three spiked concentrations of 0.1,1.0,4.0 μg/kg were in the range of 70.3%-112.5% with relative standard deviations of 2.2%-9.3%.The method was rapid,sensitive and accurate,and could be effectively applied in the rapid monitoring of 11 algal toxins in sediments.
sediments;algal toxin;ultra performance liquid chromatography-tandem mass spectrometry(UPLC-MS/MS)
O657.7;O657.63
:A
:1004-4957(2017)09-1133-06
2017-04-25;
:2017-05-24
环保部科技基金资助项目(AH20161001);安徽省环境保护科研项目(20131008)
*
:张 敏,硕士,教授级高工,研究方向:环境保护监测理论与新技术,Tel:18919635957,E-mail: 298889005@qq.com
10.3969/j.issn.1004-4957.2017.09.014
猜你喜欢
少年文艺(2022年8期)2022-07-08 10:02:47
海洋通报(2022年2期)2022-06-30 06:07:04
海洋石油(2021年3期)2021-11-05 07:43:12
考试与评价·高二版(2021年3期)2021-09-10 07:22:44
河北环境工程学院学报(2021年1期)2021-03-19 08:43:00
数学物理学报(2020年5期)2020-11-26 06:06:28
天然产物研究与开发(2018年8期)2018-09-10 05:48:38
中国经济周刊(2017年6期)2017-03-21 00:59:27
读写算·高年级(2016年3期)2016-05-30 01:53:46
公民与法治(2016年14期)2016-05-17 04:15:03
