
固相萃取-超高效液相色谱-高分辨质谱法快速测定食用油中4种酚类环境雌激素残留
2017-09-14 11:07潘胜东陈晓红邱巧丽金米聪
色谱 2017年9期
潘胜东, 何 仟, 陈晓红, 王 立, 邱巧丽, 金米聪*
(1. 宁波市疾病预防控制中心, 浙江省微量有毒化学物健康风险评估技术研究重点实验室, 宁波市毒物研究与控制重点实验室, 浙江 宁波 315010; 2. 中国疾病预防控制中心, 职业卫生与中毒控制所, 北京 100050)
研究论文
固相萃取-超高效液相色谱-高分辨质谱法快速测定食用油中4种酚类环境雌激素残留
潘胜东1, 何 仟2, 陈晓红1, 王 立1, 邱巧丽1, 金米聪1*
(1. 宁波市疾病预防控制中心, 浙江省微量有毒化学物健康风险评估技术研究重点实验室, 宁波市毒物研究与控制重点实验室, 浙江 宁波 315010; 2. 中国疾病预防控制中心, 职业卫生与中毒控制所, 北京 100050)
建立了固相萃取-超高效液相色谱-高分辨质谱(SPE-UPLC-HRMS)快速测定食用油中4种痕量酚类环境雌激素(双酚S(BPS)、双酚F(BPF)、双酚A(BPA)和4-壬基酚(4-NP))的分析方法。食用油样品经乙腈涡旋提取和SLC玻璃固相萃取小柱净化后,以0.05%(v/v)三乙醇胺和甲醇溶液为流动相进行梯度洗脱,采用Waters ACQUITY UPLC HSS T3色谱柱(100 mm×2.1 mm, 1.8 μm)进行分离,在电喷雾离子源负离子模式(ESI-)和选择性离子监测(SIM)模式下进行检测,内标法定量。4种目标物在各自的范围内有良好的线性关系,相关系数(r)>0.999。方法的检出限(LOD,S/N=3)和定量限(LOD,S/N=10)分别为0.03~0.11 μg/kg和0.10~0.36 μg/kg。在1.0、10.0和80.0 μg/kg 3个加标水平下,4种目标物的加标回收率为86.3%~96.1%,相对标准偏差(RSD)为2.2%~8.8%(n=6)。基质效应实验表明方法在低、中、高3个浓度水平下均无明显的基质干扰。该法简便、快速,能用于食用油中双酚S、双酚F、双酚A和4-壬基酚残留的快速检测。
超高效液相色谱-高分辨质谱;固相萃取;酚类环境雌激素;双酚S;双酚F;双酚A;4-壬基酚;食用油
双酚A(BPA)、双酚F(BPF)和4-壬基酚(4-NP)均为内分泌干扰物质,具有类似雌激素的作用,低剂量水平的BPA即可干扰正常的内分泌调节[1-3],过量摄入则可能影响生殖功能,甚至能引起细胞癌变和器官畸形发育[4,5]。据统计,欧洲每年消耗约69万吨BPA[6],BPA可通过多种方式进入环境,危及人类健康。随着我国经济的快速发展,BPA等内分泌干扰物的生产和需求不断增加[7]。BPA被广泛添加于食品包装材料、容器内壁涂料等材料中,塑料制品中也常含有BPA和4-NP等有害成分。因此,在塑料桶装食用油的储存过程中容器内壁的BPA等环境雌激素可能会析出,从而影响人类健康。为此,相对更安全的双酚S(BPS)作为BPA的替代物被广泛使用,然而最新研究发现,BPS可能和BPA同样有害,体外实验已证实BPS具有雌激素效应及抗雄性激素效应,其激素效应分别为BPA的32%和25%[8]。此外,实验发现长期暴露在低剂量BPS环境中的雌鼠肥胖的概率明显增加[9]。
目前,国内外针对BPA和BPS等酚类环境雌激素在食品中的检测方法主要包括气相色谱-质谱法(GC-MS)[10-12]、液相色谱-紫外可见光谱法(HPLC-UV)[13]和液相色谱-串联质谱法(LC-MS/MS)[14-17]等。GC-MS的优点在于分离度好,对双酚A和烷基酚的灵敏度要高于液相色谱法,但是酚类化合物的挥发性差,需要对目标物进行衍生化反应(硅烷化反应、苄基化反应、乙酰化反应和氟酰化反应),操作繁琐,前处理时间较长,不如液相色谱法操作简单[18]; HPLC-UV测定BPA类化合物不用衍生化,可以保持化合物的组成不变,但灵敏度较低;LC-MS/MS能有效弥补HPLC-UV的不足,具有操作简单、假阳性率低的特点,尤其适用于食品中多组分的同时检测,目前已大量用于食品安全的残留分析[19-23]。但针对食品样品的分析,采用LC-MS/MS时产生的基质效应却不可避免[24],往往需要进行有效的基质净化才能实现准确的定性、定量分析。此外,由于BPS结构中具有砜基(-SO2-),极性较强,与BPF、BPA和4-NP的色谱行为差异性大,在常规C18色谱柱上保留能力差,大大限制了复杂基质样品的测定。
本研究选择对极性化合物具有更强保留能力的Waters ACQUITY UPLC HSS T3色谱柱进行分离,采用超高效液相色谱-高分辨质谱作为检测手段,通过准分子离子峰精确的相对分子质量定性(误差<0.50 ppm (1×10-6)),开发了基于固相萃取净化模式快速检测食用油中BPS、BPF、BPA和4-NP 4种酚类环境雌激素的方法。该法快速、准确,为食品安全风险监测提供了新思路。
1 实验部分
1.1仪器和试剂
Waters UPLC I Class型超高效液相色谱仪(美国Waters公司); Q-Exactive Orbitrap型高分辨质谱仪,配有Trace Finder 3.3数据处理系统(美国Thermo Fisher公司); Legend RT型离心机(德国Heraeus公司); SLC玻璃固相萃取小柱(500 mg/6 mL,杭州福裕科技服务有限公司)。
甲醇(LC-MS级,批号:20160205)购自美国Thermo Fisher公司;氨水(色谱纯,批号:83Y1409PF)和三乙醇胺(色谱纯,批号:81Y1509PF)购自德国Merck公司;BPS(批号:127044)、BPF(批号:50107)、BPA(批号:40217)、4-NP(批号:90819)标准品和13C12-BPS、D4-BPA同位素内标均购自德国Dr. Ehrenstorfer公司。食用油样品购自各大型超市。
1.2标准溶液的配制
分别准确称取1.0 mg BPS、BPF、BPA和4-NP于1.0 mL容量瓶中,用甲醇溶解并定容至刻度,配制质量浓度为1 000 mg/L的4种目标物的标准储备液;分别准确移取上述4种标准储备液100 μL,置于100 mL容量瓶中,用甲醇稀释并定容至刻度,配制质量浓度为1.0 mg/L的混合标准溶液。
分别准确称取1.0 mg13C12-BPS和D4-BPA,置于1.0 mL容量瓶中,用甲醇溶解并定容至刻度,配制质量浓度为1 000 mg/L的内标储备液;分别准确移取上述2种内标储备液100 μL,置于100 mL容量瓶中,用甲醇稀释并定容至刻度,配制质量浓度为1.0 mg/L的内标应用液。
分别准确吸取1.0、2.0、5.0、10.0、25.0、100.0 μL 1.0 mg/L的混合标准溶液,置于10.0 mL容量瓶中,分别加入50.0 μL 1.0 mg/L的内标应用液,用甲醇稀释并定容至刻度,配制质量浓度分别为0.1、0.2、0.5、1.0、5.0、10.0、25.0、100.0 μg/L的系列混合标准溶液,其中内标13C12-BPS和D4-BPA的质量浓度均为5.0 μg/L。
1.3样品前处理
提取:称取0.50 g(精确至0.01 g)食用植物油,置于10 mL具塞玻璃刻度试管,加入5.0 μL 1.0 mg/L的内标应用液,混匀,加入2 mL乙腈,涡旋混匀5 min,以10 000 r/min离心2 min,取上层乙腈相,待净化。
净化:依次用5 mL二氯甲烷和5 mL乙腈活化SLC玻璃固相萃取小柱,然后移取2 mL上述提取液,上样,收集流出液于试管中,再用2 mL乙腈洗脱,并收集洗脱液于同一试管中,于60 ℃氮气吹至近干,用1 mL甲醇复溶,进样测定。
1.4色谱条件
色谱柱:Waters ACQUITY UPLC HSS T3柱(100 mm×2.1 mm, 1.8 μm);流速:0.3 mL/min;进样量:5.0 μL;流动相:A相为0.05%(v/v)三乙醇胺溶液,B相为甲醇。梯度洗脱程序:0~4.00 min, 20%B~95%B; 4.00~5.00 min, 95%B; 5.00~5.01 min, 95%B~20%B; 5.01~7.00 min, 20%B。
1.5质谱条件
离子源:电喷雾离子(ESI)源;扫描方式:负离子扫描;离子源温度:320 ℃;定量检测方式:选择性离子监测模式(SIM);电喷雾电压:-2.8 kV;溶剂化气压力:275.8 kPa;辅助气速率:180 L/h;射频棱镜电压(S-lens RF level): 60%;辅助气加热温度:300 ℃;分辨率:35 000;自动增益控制(AGC): 1×105;最大注入时间(maximum IT): 60 ms。4种酚类环境雌激素及2种内标物的准分子离子峰均为[M-H]-,分别以各化合物准分子离子峰的精确相对分子质量(Mr)和保留时间(tR)定性,内标法定量。表1列举了BPS、BPF、BPA和4-NP及内标物的质谱参数。

表 1 BPS、BPF、BPA和4-NP及内标物的分子式、相对分子质量、质量数偏差和保留时间Table 1 Formulas, relative molecular mass (Mr), mass deviations and retention times (tR) of BPS, BPF, BPA, 4-NP and their internal standards
2 结果与讨论
2.1色谱条件的优化

图 1 不同色谱柱与流动相条件下BPS(5.0 μg/L)的色谱图Fig. 1 Chromatograms of BPS (5.0 μg/L) with different kinds of chromatographic columns and mobile phases a. column: Waters BEH C18 column (100 mm×2.1 mm, 1.7 μm); mobile phases: 0.1% (v/v) ammonia-methanol; b. column: Waters BEH C18 column (100 mm×2.1 mm, 1.8 μm); mobile phases: 0.05% (v/v) triethanolamine-methanol; c. column: Waters HSS T3 column (100 mm×2.1 mm, 1.8 μm); mobile phases: 0.1% (v/v) ammonia-methanol; d. column: Waters HSS T3 column (100 mm×2.1 mm, 1.8 μm); mobile phases: 0.05% (v/v) triethanolamine-methanol.
采用液相色谱分离分析有机化合物时,C18色谱柱是首选,本研究首先采用Waters BEH C18色谱柱(100 mm×2.1 mm, 1.7 μm)分离BPS、BPF、BPA和4-NP 4种酚类环境雌激素,并分别考察了水-甲醇、乙酸铵-甲醇、0.1%(v/v)氨水-甲醇和0.05%(v/v)三乙醇胺-甲醇作为流动相时4种酚类环境雌激素的分离效果和灵敏度。结果表明,当水-甲醇作为流动相时,4种酚类环境雌激素能较好地分离,BPS的保留也较好,但BPA的灵敏度较差,不能满足痕量检测的要求,且4-NP的保留时间滞后严重,峰形不佳;当乙酸铵-甲醇溶液作为流动相时,4-NP的保留时间合适,但BPA基本无质谱响应;当0.1%(v/v)氨水-甲醇作为流动相时,4种待测物能较好地实现分离,峰形对称,灵敏度好,但BPS的保留较差(tR=0.57 min),接近死时间(见图1a);当选用0.05%(v/v)三乙醇胺-甲醇作为流动相时,BPS的保留有所改善,tR为1.04 min(见图1b),可能是因为BPS分子中含有-SO2-和-OH基团,极性较大,在碱性条件下,电离成负离子,较难在C18色谱柱上保留,而三乙醇胺的存在可能与BPS通过氢键作用力形成复合物,从而增强了在C18柱上的保留,但由于BPS的保留时间依然较短,有待进一步完善。之后选用对极性化合物具有更强保留能力的Waters HSS T3色谱柱(100 mm×2.1 mm, 1.8 μm)进行分析。当水-甲醇和乙酸铵-甲醇作为流动相时,各目标化合物的色谱和质谱行为与使用Waters BEH C18色谱柱(100 mm×2.1 mm, 1.7 μm)时相似,BPA质谱响应较弱,不能满足残留分析的要求;当0.1% (v/v)氨水-甲醇作为流动相时,BPS在T3色谱柱上的保留依然较差(tR=0.92 min,见图1c);当以0.05% (v/v)三乙醇胺-甲醇作为流动相进行分离时,BPS在色谱柱上的保留能力有了较大提高,保留时间为1.62 min(见图1d),且此时BPA、BPF和4-NP的保留、峰形和灵敏度也较好。综上,本文采用Waters HSS T3色谱柱进行分离,选择0.05% (v/v)三乙醇胺-甲醇作为流动相进行梯度洗脱。加标样品中4种酚类环境雌激素及2种内标物的SIM色谱图见图2。
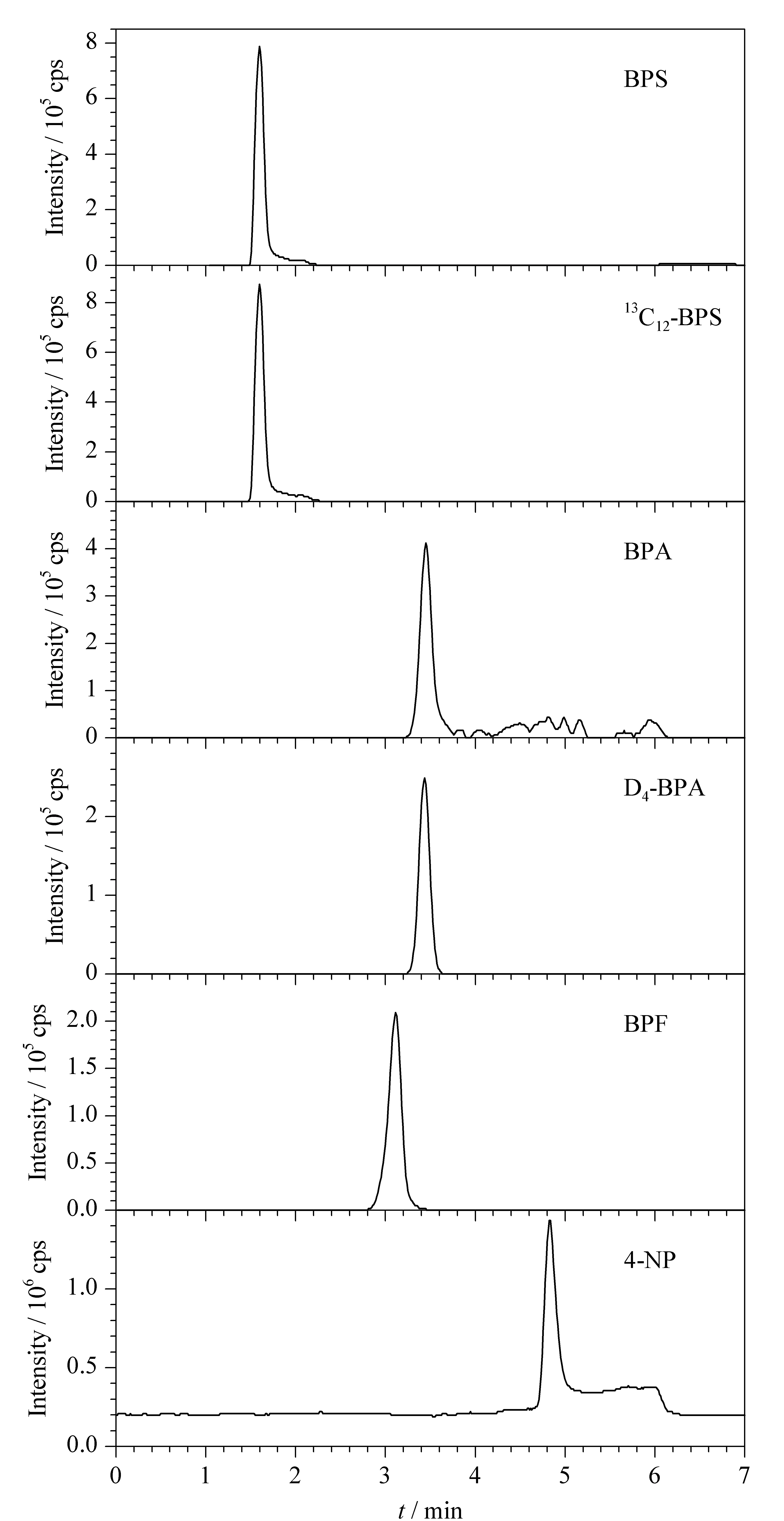
图 2 加标食用油样品中4种酚类环境雌激素及2种内标物(5.0 μg/kg)的SIM色谱图Fig. 2 SIM chromatograms of the four phenolic environmental estrogens and two internal standards (5.0 μg/kg) spiked in cooking oil samples
2.2质谱条件的优化
由于本文所研究的4种酚类环境雌激素的分子结构中均含有羟基,容易电离形成负离子,因此在ESI-模式下对各目标化合物进行分析。实验表明,当采用一级质谱时,在ESI-模式下,4种酚类环境雌激素所产生的准分子离子均为[M-H]-,鉴于通过高分辨质谱获得的精确相对分子质量与理论值间偏差较小(均<0.50 ppm,见表1),准分子离子完全可以作为各化合物定性分析的依据。因此,本文采用Targeted SIM模式对4种酚类环境雌激素进行定性定量分析,并对影响Q-Exactive灵敏度和选择性的各参数(如分辨率、自动增益控制、最大注入时间)进行优化,结果表明,当设定分辨率为35 000、AGC为1×105和最大注入时间为60 ms时,4种酚类环境雌激素及其内标物的质谱响应程度最强,选择性最佳。优化后的质谱参数见表1。
2.3前处理条件的优化
2.3.1提取溶剂的选择
本研究分析的4种酚类环境雌激素的化学结构中均带有一个或多个羟基极性基团,根据相似相溶原理,其在极性溶剂中的溶解度应较为理想。因此本实验考察了甲醇、乙腈和丙酮3种溶剂对目标分析物的提取效率。实验结果表明,当采用乙腈提取时,食用油基质中的待测物可被充分提取,4种酚类环境雌激素的提取率为93.6%~99.5%,优于其他两种提取溶剂。因此本实验选择乙腈作为食用油中酚类环境雌激素的提取溶剂。
2.3.2净化方式的选择
食用油样品基质较为复杂,尤其含有大量的脂肪,导致采用质谱分析时基质效应显著,需对提取液进行适当的净化。然而,酚类环境雌激素在塑料制品中普遍残留,常规的塑料固相萃取小柱可能会对实验结果造成影响,因此选择分散固相萃取或玻璃填充固相萃取小柱能有效减少不必要的干扰。

图 3 不同净化方式对4种酚类环境雌激素回收率的影响(n=3)Fig. 3 Effect of different clean-up approaches on the recoveries of the four phenolic environmental estrogens (n=3)
本研究分别比较了采用C18粉末、SILICA/PSA玻璃混合型固相萃取小柱和SLC玻璃固相萃取小柱时目标分析物的回收率(见图3)。实验结果表明,C18粉末能有效去除食用油中的非极性杂质,BPS、BPA和4-NP的回收率可达76.5%~86.5%,但BPF的回收率较低(<65%); 采用SILICA/PSA玻璃混合型固相萃取小柱时,BPF、BPA和4-NP的回收率为85.9%~92.6%,但BPS的保留较强,几乎难以洗脱,回收率<15%;SLC玻璃固相萃取小柱由中性氧化铝和强阳离子交换材料(SCX)复合而成,在净化过程中,食用油样品经乙腈提取后,提取液中含有一定量的甘油三酯,可通过疏水作用机理吸附于氧化铝表面,从而达到除杂的目的,而SCX成分可通过静电作用机理有效吸附提取液中离子型杂质,进而实现杂质净化的目的。实验结果表明,4种酚类环境雌激素的回收率为93.1%~98.8%,因此实验选用SLC玻璃固相萃取小柱进行净化。
2.4基质效应的评价
在不含BPS、BPF、BPA和4-NP的食用油提取液(经固相萃取柱净化)中分别添加低、中、高3个水平(1.0、10.0和80.0 μg/kg)的BPS、BPF、BPA和4-NP,比较相同水平下食用油基质中目标物峰面积(S2)与标准品溶液中目标物峰面积(S1)间的差异,并计算绝对基质效应(Absolute matrix effect, AME),公式为:AME=S2/S1×100%。实验结果表明,BPS、BPF、BPA和4-NP在低、中、高3个添加水平下的绝对基质效应为84.3%~96.3%,表明食用油样品经SLC固相萃取净化后的基质干扰不明显,提取净化效果较好。
2.5方法学验证
对配制的质量浓度为0.1~100.0 μg/L的系列标准溶液进行分析,根据内标物与被测物的性质,选择13C12-BPS作为BPS的内标物、D4-BPA作为BPF、BPA和4-NP的内标物,以被测组分与内标物的峰面积之比(y)对标准溶液中被测组分与内标物的质量浓度之比(x)进行线性回归,结果见表2。由表2可知,4种酚类环境雌激素在各自的范围内均具有良好的线性关系,相关系数(r)>0.999。采用空白食用油样品中添加待测物的方式确定方法的检出限(LOD)和定量限(LOQ),分别以目标分析物色谱峰信噪比(S/N)为3和10时计算,4种酚类环境雌激素的LOD和LOQ分别为0.03~0.11 μg/kg和0.10~0.36 μg/kg。
准确称取18份0.50 g(精确至0.01 g)空白食用油样品,置于10 mL具塞玻璃刻度试管中,以6份为一组,加入5.0 μL 1.0 mg/L的内标应用液,然后分别准确加入1.0 mg/L的混合标准溶液各1.0、10.0和80.0 μL,配制低、中、高3个添加水平的加标样品(1.0, 10.0和80.0 μg/kg),按1.3节进行前处理,然后采用UPLC-HRMS测定,每一水平平行测定6次,计算回收率和精密度(见表2)。结果表明,食用油样品中4种目标分析物的加标回收率为86.3%~96.1%,RSD为2.2%~8.8%(n=6)。
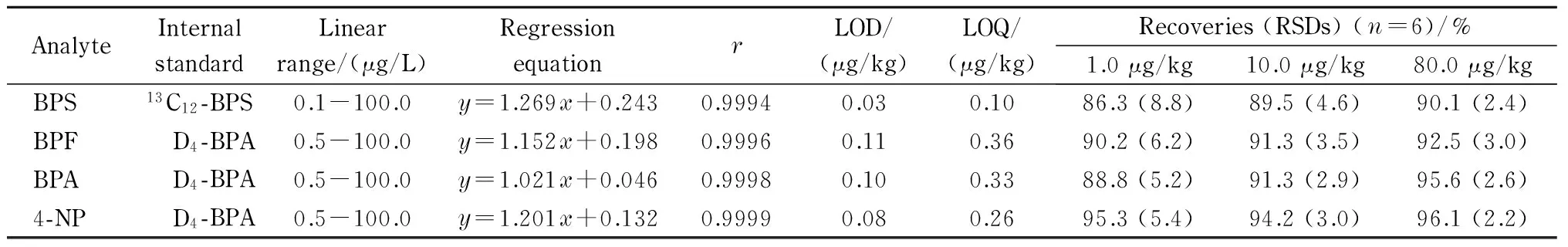
表 2 4种酚类环境雌激素的线性范围、回归方程、相关系数、检出限、定量限、回收率和精密度Table 2 Linear ranges, regression equations, correlation coefficients (r), LODs, LOQs, recoveries, and precisions of the four phenolic environmental estrogens
y: peak area ratio of target analyte to its internal standard;x: mass concentration ratio of target analyte to its internal standard.
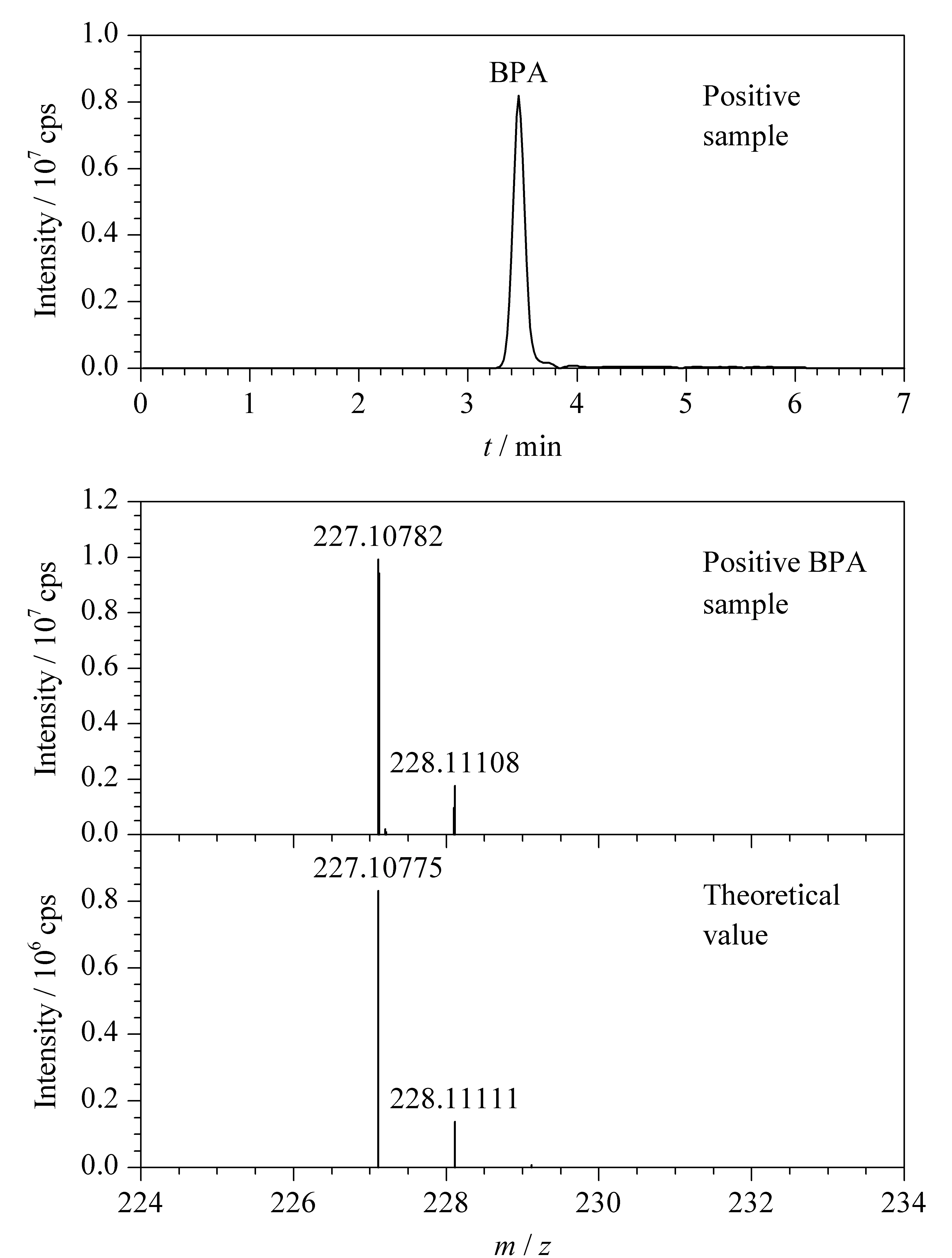
图 4 典型阳性样品中BPA的SIM色谱图及其一级质谱图和理论质谱图Fig. 4 SIM chromatogram of a BPA positive sample and its experimental and theoretical mass spectra
2.6实际样品分析
采集市售食用油样品30份作为研究对象,采用本文建立的SPE-UPLC-MS/MS方法测定4种酚类环境雌激素。实验结果表明,共检出15份食用油样品中含有BPA, 10份含有BPS, 5份含有4-NP, 2份含有BPF,总体的含量范围为1.1~50.2 μg/kg。典型的BPA阳性样品的SIM色谱图和质谱图见图4。由图4可知,BPA阳性样品一级质谱图与BPA的理论图谱极其接近,两个主要的准分子离子同位素峰的精确相对分子质量的偏差均<0.50 ppm,定性能力好。由此可见,本研究所建立的食用油中酚类环境雌激素的检测方法具有准确可靠的优点。
3 结论
本文建立了固相萃取-超高效液相色谱-高分辨质谱测定食用油中4种痕量酚类环境雌激素残留的分析方法。该法具有快速、准确、灵敏和定性能力好等优点,可用于食用油中酚类环境雌激素的快速确证分析。
[1] Jocelyn K. Science, 2000, 290(5492): 695
[2] Lemos M F L, van Gestel C A M, Soares A M V M. J Soil Sediment, 2010, 9(5): 492
[3] Hecker M, Hollert H. Environ Sci Europe, 2011, 23(1): 15
[4] Vandenberg L N, Maffini M V, Sonnenschein C, et al. Endocr Rev, 2009, 30(1): 75
[5] Li Z D, Han Z Y, Guo G R. Thermosetting Resin, 2011, 26(4): 50
李振东, 韩占元, 郭桂荣. 热固性树脂, 2011, 26(4): 50
[6] European Union. European Union Risk Assessment Report-BPA (4,4′-isopropylidenediphenol (Bisphenol-A)). Luxembourg: Office for Official Publications of the European Communities, 2008
[7] Shao B, Han H, Li D M, et al. Chinese Journal of Chromatography, 2005, 23(4): 362
邵兵, 韩灏, 李冬梅, 等. 色谱, 2005, 23(4): 362
[8] Del Moral L I, Le Corre L, Poirier H, et al. Toxicology, 2016, 357/358(1): 11
[9] Rochester J R, Bolden A L. Environ Health Persp, 2015, 123(7): 643
[10] Du H F, Yan H F, Li Y, et al. Journal of Hygiene Research, 2007, 36(3): 369
杜会芳, 闫慧芳, 李晔, 等. 卫生研究, 2007, 36(3): 369
[11] Zhang X Z, Xu Y J, Gong X H, et al. Journal of Fishery Sciences of China, 2009, 16(5): 791
张秀珍, 徐英江, 宫向红, 等. 中国水产科学, 2009, 16(5): 791
[12] Jia Y Y, Tan J H, Xu C, et al. Chinese Journal of Chromatography, 2014, 32(3): 263
贾妍艳, 谭建华, 徐晨, 等. 色谱, 2014, 32(3): 263
[13] Zhang L, Li N, Wan Y Y. The Administration and Technique of Environmental Monitoring, 2010, 22(1): 49
张丽, 李楠, 万延延. 环境监测管理与技术, 2010, 22(1): 49
[14] Liao C Y, Kannan K. J Agric Food Chem, 2013, 61(19): 4655
[15] Niu Y M, Zhang J, Zhang S J, et al. Chinese Journal of Analytical Chemistry, 2012, 40(4): 534
牛宇敏, 张晶, 张书军, 等. 分析化学, 2012, 40(4): 534
[16] Ding J, Zhang S H, Liu J N, et al. Chinese Journal of Chromatography, 2014, 32(5): 529
丁洁, 张圣虎, 刘济宁, 等. 色谱, 2014, 32(5): 529
[17] Kou L J, Liang R N. Chinese Journal of Chromatography, 2014, 32(8): 817
寇立娟, 梁荣宁. 色谱, 2014, 32(8): 817
[18] Huang H, Gong X H, Deng X X, et al. Chinese Fishery Quality and Standards, 2011, 1(3): 62
黄会, 宫向红, 邓旭修, 等. 中国渔业质量与标准, 2011, 1(3): 62
[19] Wang S, Zhang J, Shao B. Journal of Instrumental Analysis, 2013, 32(2): 179
王硕, 张晶, 邵兵. 分析测试学报, 2013, 32(2): 179
[20] Chen X H, Zhu H, Zhou L X, et al. Journal of Hygiene Research, 2014, 43(3): 455
陈晓红, 朱浩, 周丽新, 等. 卫生研究, 2014, 43(3): 455
[21] Pan S D, Zhao Y G, Chen X H, et al. Chinese Journal of Health Laboratory Technology, 2014, 24(1): 24
潘胜东, 赵永纲, 陈晓红, 等. 中国卫生检验杂志, 2014, 24(1): 24
[22] Hu Z Y, Wu P G, Wang T J, et al. Journal of Health Laboratory Technology, 2016, 26(21): 3083
胡争艳, 吴平谷, 王天娇, 等. 中国卫生检验杂志, 2016, 26(21): 3083
[23] Pan S D, Li X P, Chen X H, et al. Journal of Health Laboratory Technology, 2016, 26(8): 1072
潘胜东, 李小平, 陈晓红, 等. 中国卫生检验杂志, 2016, 26(8): 1072
[24] Cui C Y, Zhang H Y, Wu X Q, et al. Chinese Journal of Chromatography, 2017, 35(5): 487
崔春艳, 张红医, 吴兴强, 等. 色谱, 2017, 35(5): 487
Rapid determination of four phenolic environmental estrogen residues in cooking oil by ultra-performance liquid chromatography-high resolution mass spectrometry coupled with solid-phase extraction
PAN Shengdong1, HE Qian2, CHEN Xiaohong1, WANG Li1, QIU Qiaoli1, JIN Micong1*
(1. Zhejiang Provincial Key Laboratory of Health Risk Appraisal for Trace Toxic Chemicals, Ningbo Key Laboratory of Poison Research and Control, Ningbo Municipal Center for Disease Control and Prevention,Ningbo 315010, China; 2. National Institute of Occupational Health and Poison Control, Chinese Center for Disease Control and Prevention, Beijing 100050, China)
A fast, sensitive and accurate method for the determination of trace bisphenol S (BPS), bisphenol F (BPF), bisphenol A (BPA) and 4-nonylphenol (4-NP) in cooking oil samples was developed by ultra-performance liquid chromatography-high resolution mass spectrometry (UPLC-HRMS) coupled with solid-phase extraction (SPE). Cooking oil samples were extracted by acetonitrile, then the supernatant was purified by SLC SPE cartridges. The chromatographic separation was carried out on a Waters ACQUITY UPLC HSS T3column (100 mm×2.1 mm, 1.8 μm) with a linear gradient elution procedure using 0.05% (v/v) triethanolamine aqueous solution and methanol as mobile phases. The quantification analysis was operated in a negative electrospray ion (ESI-) source mode under the selected ion monitoring (SIM) mode with internal standard method. The four target analytes showed good linearity with correlation coefficients (r) greater than 0.999. The limits of detection (LODs,S/N=3) and limits of quantification (LOQs,S/N=10) were in the ranges of 0.03-0.11 μg/kg and 0.10-0.36 μg/kg, respectively. The recoveries of the four target analytes spiked in oil samples were in the range of 86.3%-96.1% at spiked levels of 1.0, 10.0 and 80.0 μg/kg, respectively, while the relative standard deviations (RSDs) were in range of 2.2%-8.8% (n=6). No significant matrix interference was found in this method. The proposed method is simple and fast. It can be applied for the rapid determination of trace BPS, BPF, BPA, and 4-NP in cooking oil samples.
ultra-performance liquid chromatography-high resolution mass spectrometry (UPLC-HRMS); solid-phase extraction (SPE); phenolic environmental estrogens; bisphenol S (BPS); bisphenol F (BPF); bisphenol A (BPA); 4-nonylphenol (4-NP); cooking oil
10.3724/SP.J.1123.2017.06003
2017-06-01
.E-mail: jmcjc@163.com.
浙江省医药卫生平台骨干项目(2015RCB023);浙江省医药卫生平台重点项目(2014ZDA021);宁波市自然科学基金项目(2016A610178);浙江省公共卫生应急检测关键技术重点实验室开放基金.
O658
A
1000-8713(2017)09-0980-07
Foundation item: Zhejiang Provincial Medical Health Platform Backbone Project (No. 2015RCB023); Zhejiang Provincial Medical Health Platform Key Project (No. 2014ZDA021); Ningbo Natural Science Foundation of China (No. 2016A610178); Opening Foundation of Key Laboratory of Key Technologies of Emergency Detection for Public Health of Zhejiang Province.
*
猜你喜欢
少年文艺(2022年8期)2022-07-08
天然产物研究与开发(2018年9期)2018-10-08
中成药(2017年7期)2017-11-22
中成药(2017年3期)2017-05-17
中国经济周刊(2017年6期)2017-03-21
中国塑料(2016年4期)2016-06-27
中国塑料(2016年2期)2016-06-15
中国医疗美容(2015年1期)2015-07-12
食品工业科技(2014年13期)2014-03-11
中国医学科学院学报(2013年6期)2013-03-11
- 色谱的其它文章
- 气相色谱法测定及预测有机磷阻燃剂的蒸气压
- 超分子溶剂直提测定水中的多环芳烃
- Separation and purification of acteoside from Rehmanniaglutinosa by combining macroporous resin with high-speed countercurrent chromatography
- Preparation of filter paper with chiral separation function by oxidation and Schiff-base reaction
- 超高效液相色谱-串联质谱法检测水果中6种甲氧基丙烯酸酯类杀菌剂及其质谱裂解规律
- 《色谱》论文中可直接使用的缩略词