
邻异丙硫基苯甲酰基和苯硫甲基铁衍生物的合成与反应
2017-09-12 08:59:35赵达维徐越孙宝川王志宏唐良富
无机化学学报 2017年9期
赵达维徐越孙宝川王志宏唐良富
邻异丙硫基苯甲酰基和苯硫甲基铁衍生物的合成与反应
赵达维徐越孙宝川王志宏唐良富*
(南开大学化学学院,元素有机化学国家重点实验室,天津300071)
利用异丙基苯硫醚与丁基锂反应后,再依次与羰基铁和碘反应制得了碘桥双核邻异丙硫基苯甲酰基铁配合物[(o-iPrS) C6H4COFe(CO)2I]2,而苯甲硫醚类似的反应却仅得到单核苯硫甲基铁配合物C6H5SCH2Fe(CO)3I。当与亲核试剂作用时,这2个化合物表现出显著不同的反应活性。如双核配合物[(o-iPrS)C6H4COFe(CO)2I]2与2-吡啶硫醇钠(PySNa)反应得到单核配合物(o-iPrS) C6H4COFe(CO)2(SPy),但单核配合物C6H5SCH2Fe(CO)3I与PySNa反应导致其分解。另一方面,单核配合物C6H5SCH2Fe(CO)3I与三苯基膦(PPh3)反应得到羰基取代配合物C6H5SCH2Fe(CO)2(PPh3)I,但是双核配合物[(o-iPrS)C6H4COFe(CO)2I]2类似的反应却导致其分解,没有获得可表征的化合物。所有新合成的化合物都通过了核磁与红外光谱的表征,它们的结构也获得了X射线单晶衍射的确证。
硫醚;羰基铁;金属酰基配合物;吡啶;膦配体
The investigation on the metal-acyl complexes continues to be an active research area in organometallic chemistry since these complexes show excellent catalytic activity in many organic transformations[1-4], and this category of complexes are proposed as the key active intermediates in numerous catalytic processes[5-8]. Recent studies have proven the practicability that the metal-acyl moiety was introduced into the main chain of polymers to form potential functional organometallic materials[9],and the organometallic aqueous colloids were formed through the self-assembly of the metalacyl complexes[10],whichmarkedly broaden their application fields.Among the metal-acyl complexes, the acyl iron complexes have drawn more and more attention in recent years because of the successful elucidation of the structure of[Fe]-hydrogenase[11-13], which suggests that an acyl-iron ligates to the active site of[Fe]-hydrogenase[14].Since then a number of acyl iron complexes have been synthesized and characterized[15-20],which promotes the rapid development of acyl iron complexes.To match the electronic and steric properties of biomimetic models for the active site of [Fe]-hydrogenase,several kinds of donor-functionalized acyl ligands,such as acylphosphine[21],carbamoylpyridine[19],acylmethylpyridine[16],andacylmethylpyrazole[22-23],have been used to synthesize acyl iron complexes.Because of the introduction of the chelationassisted donor atoms bearing different electron-donating ability,the corresponding acyl iron complexes exhibit versatile reactivity.In addition,the incorporation of the sulfur-containing ligand to the metal center of model complexes for[Fe]-hydrogenase is also important, since the sulfur atom possibly plays a role in the catalytic cycle of hydrogen activation by[Fe]-hydrogenase[24-25].However,iron complexes with sulfurfunctionalized acyl ligand[26-27]are rare in known synthetic model complexes of[Fe]-hydrogenase,in which phenylthiolate or 2-pyridinethiolate ligand is usually used to occupy the cysteine sulfur position.Other structuralmodelswithsulfur-functionalizedacyl ligand should be conducive to gaining a deeper understanding of the structure and catalytic function of[Fe]-hydrogenase owing to the strong influence of the stereo-electronic environment of the iron center on the ability of the biomimic to bind hydrogen molecule. Our previous investigations showed that the different chelation-assisted donor atoms significantly affected the structure and reactivity of the corresponding acyl iron complexes[22-23].As an extension of our investigations on this subject,herein we describe the synthesis and reactivity of thioether-based iron complexes.
1 Experimental
Solvents were dried and freshly distilled prior to use according to standard procedures.All reactions were carried out under an atmosphere of argon.NMR spectra were recorded on a Bruker 400 spectrometer using CDCl3as solvent unless otherwise noted,and the chemical shifts were reported with respect to the reference(internal SiMe4for1H and13C NMR spectra, external 85%H3PO4aqueous solution for31P NMR spectra).IR spectra were recorded as KBr pellets on a Tensor 27 spectrometer.Elemental analyses were carried out on an Elementar Vario EL analyzer.
1.1 Synthesis of[(o-iPrS)C6H4COFe(CO)2I]2(1)
A hexane solution of n-BuLi(1.6 mol·L-1,1.3 mL,2 mmol)was added to the solution of N,N,N′,N′-tetramethylethylenediamine(TMEDA)(0.2 mL,2 mmol)in hexane(10 mL)at room temperature.After 30 min,isopropylthiobenzene(0.32 mL,2 mmol)was added to the above-mentioned solution.The resulting mixture was stirred for 4 h at room temperature,during which time a solid was precipitated out.Then,the reaction mixture was cooled to-78℃,and THF(20 mL)was added,After the solid was completely dissolved, Fe(CO)5(0.26 mL,2 mmol)was added.The reaction mixture was continuously stirred at-78℃for 30 min, and allowed to reach room temperature slowly and stirred for 2 h.A solution of I2(0.51 g,2 mmol)in THF(10 mL)was added dropwise.After completion of addition,the reaction mixture was stirred overnight. The solvent was removed under reduced pressure,and the residue was purified by column chromatography on silica with CH2Cl2as the eluent.The red eluate was concentrated to dryness to give 1 as a red solid.Yield: 0.33 g(38%).1H NMR:δ 1.43(d,J=5.3 Hz,6H,CH3),1.49(d,J=5.0 Hz,6H,CH3),3.20~3.35(m,2H,CH), 7.55~7.65(m,4H,C6H4),7.74~7.80(m,4H,C6H4).13C NMR:δ 22.1(CH3),22.4(CH3),48.7(CH),125.2,130.8, 131.7,132.1,136.4,150.5(C6H4),195.8,205.0(C≡O), 246.9(C=O).IR(cm-1):ν(C≡O)2 090,2 030,2 004, 1 975;ν(C=O)1 628.Anal.Calcd.for C24H22Fe2I2O6S2(%):C 34.48,H 2.65;Found(%):C 34.35,H 3.10.
1.2 Synthesis of(o-iPrS)C6H4COFe(CO)2(SPy)(2)
Sodium hydride(12 mg,0.5 mmol)was added to a solution of 2-mercaptopyridine(56 mg,0.5 mmol)in CH2Cl2(20 mL).The reaction mixture was stirred for 1 h at room temperature,followed by the addition of a solution of 1(0.21 g,0.25 mmol)in CH2Cl2(10 mL). The reaction mixture was continuously stirred for 6 h. The solvent was removed under reduced pressure,and the residue was purified by column chromatography on silica with CH2Cl2as the eluent.The red eluate was concentrated to dryness to afford 0.16 g(72%)of 2.This complex consisted of two isomers(2A/2B)in solution. The relative ratio of isomers was ca.1∶1 in CDCl3and 2∶1 in acetone-d6,respectively,according to the integral of the corresponding protons of the methine and methyl groups.2A:1H NMR:δ 1.38(d,J=6.6 Hz,CH3),1.49 (d,J=6.6 Hz,CH3),3.35~3.46(m,CH),3.50~3.66(m, CH),6.55~6.61(m),6.82~6.90(m),7.00~7.16(m),7.29~7.35(m),7.36~7.41(m),7.61~7.70(m),7.88~7.95(m) (phenyl and pyridyl protons).13C NMR:δ 21.5,22.0, 22.1,22.6(CH3),46.3,46.9(CH),117.5,117.9,122.2, 122.5,125.4,127.4,130.3,130.5,130.8,131.4,131.7, 131.8,135.5,136.1,138.1,138.6,148.5,148.7,149.7, 151.0,177.3,180.8(phenyl and pyridyl carbons),208.8, 210.2,210.7,211.6(C≡O),260.3,262.2(C=O).2B:1H NMR(acetone-d6):δ 1.39(d,J=6.8 Hz),1.41(d,J= 6.7 Hz),1.53(d,J=6.7 Hz),3.55~3.65(m),3.68~3.83 (m),6.77(t,J=6.2 Hz),6.89(dd,J1=2.8 Hz,J2=8.3 Hz), 7.00~7.03(m),7.09~7.24(m),7.42(dd,J1=1.0 Hz,J2= 7.6 Hz),7.89(t,J=8.1 Hz),8.20(d,J=5.3 Hz).The other signals in the range of 7.48~7.70 overlapped seriously. IR(cm-1):ν(C≡O)2 023,1 967,1 957;ν(C=O)1 615. Anal.Calcd.for C17H15FeNO3S2(%):C 50.88,H 3.77, N 3.49;Found(%):C 50.66,H 4.23,N 3.46.
1.3 Synthesis of C6H5SCH2Fe(CO)3I(3)
Thiscomplexwassimilarlyobtainedusing thioanisole instead of isopropylthiobenzene as abovementioned synthesis of 1.Yield:39%.1H NMR:δ 3.95~4.00(m,2H,CH2),7.39(s,br,3H,C6H5),7.53(s,br, 2H,C6H5).13C NMR:δ 38.7(CH2),129.8,130.0,130.1, 135.8(C6H5),203.8,209.4,210.2(C≡O).IR:ν(C≡O) 2 077,2 019,1 996 cm-1.Anal.Calcd.for C10H7FeIO3S (%):C 30.80,H 1.81;Found(%):C 30.92,H 2.14.
1.4 Synthesis of C6H5SCH2Fe(CO)2(PPh3)I(4)
PPh3(26 mg,0.1 mmol)was added to a solution of 3(39 mg,0.1 mmol)in THF(20 mL).The reaction mixture was stirred overnight at room temperature. The solvent was removed under reduced pressure,and the residue was purified by column chromatography on silica with CH2Cl2/hexane(1∶2,V/V)as the eluent to give a red eluate,which was concentrated to dryness to afford 20 mg(32%)of 4 as a red solid.1H NMR:δ 3.92~4.11(m,2H,CH2),7.03~7.60(m,20H,C6H5).13C NMR:δ 36.0(d,JP-C=20.2 Hz,CH2),128.4(d,JP-C= 9.5 Hz),128.1,129.0,129.3,130.2,133.6,133.9(d,JP-C=10.1 Hz),135.0,135.4,136.0(C6H5),214.9(d,JP-C= 17.5 Hz),218.1(C≡O).31P NMR:δ 40.1.IR:ν(C≡O)2 004,1 950 cm-1.Anal.Calcd.for C27H22FeIO2PS (%):C 51.95,H 3.55;Found(%):C 51.80,H 3.89.
1.5 Reaction of 1 with PPh3
Reaction of 1 with PPh3,like that of 3 with PPh3, resulted in the decomposition of the starting material. No characterizable product was obtained.
1.6 Crystal structure determination
Crystals of 1,2A,3 and 4 suitable for X-ray analyses were obtained by slow diffusion of hexane into their CH2Cl2solutions at-18℃.All intensity data were collected on a Rigaku Saturn CCD detector using Mo Kαradiation(λ=0.071 073 nm).Semi-empirical absorption corrections were applied using the Crystalclear program[28].The structures were solved by direct methods and difference Fourier map using SHELXS of the SHELXTL package and refined with SHELXL[29]by full-matrix least-squares on F2.All non-hydrogen atoms were refined anisotropically.Hydrogen atoms were added geometrically and refined with riding modelpositionparameters.Asummaryofthe fundamental crystal data for 1,2A,3 and 4 is listed in Table 1.
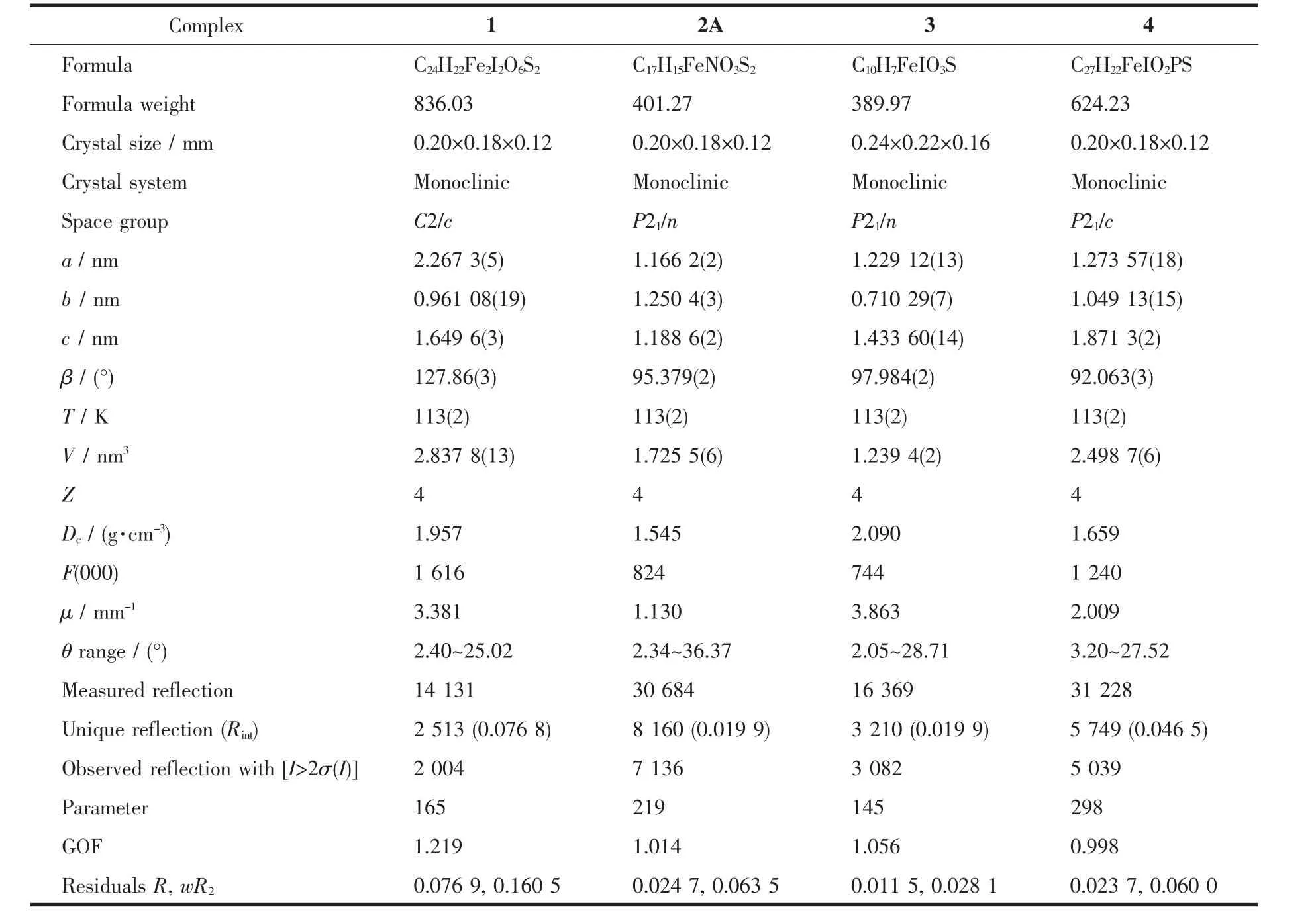
Table1 Crystal data and refinement parameters for complexes 1,2A,3 and 4
CCDC:1545834,1;1545835,2A;1545836,3; 1545837,4.
2 Results and discussion
2.1 Synthesis and reactivity of ortho-isopropylthiobenzoyl iron complex
It is known that isopropylthiobenzene is deprotonated with n-BuLi in the presence of TMEDA to give the ortho-lithiation product[30].Herein,we find that reaction of this ortho-lithiation product with iron carbonyl followed by iodine yields a dimeric orthoisopropylthiobenzoyl iron complex 1(Scheme 1).This result is significantly different from the similar reactions of(pyrazol-1-yl)methyllithium[23]and(2-pyridyl) methyllithium[31],which only give mononuclear acyl iron complexes,revealing that different chelation-assisteddonors strongly affect the fundamental structures of metal-acyl complexes.The substitution of iodide in 1 by 2-pyridinethiolate is successful,which gives mononuclear complex 2.However,reaction of 1 with PPh3leads to the decomposition of the starting material. Complexes 1 and 2 are air-stable solids and their solution could be manipulated in air for a short time withoutnotabledecomposition.Theyhavebeen characterized by spectroscopic methods.
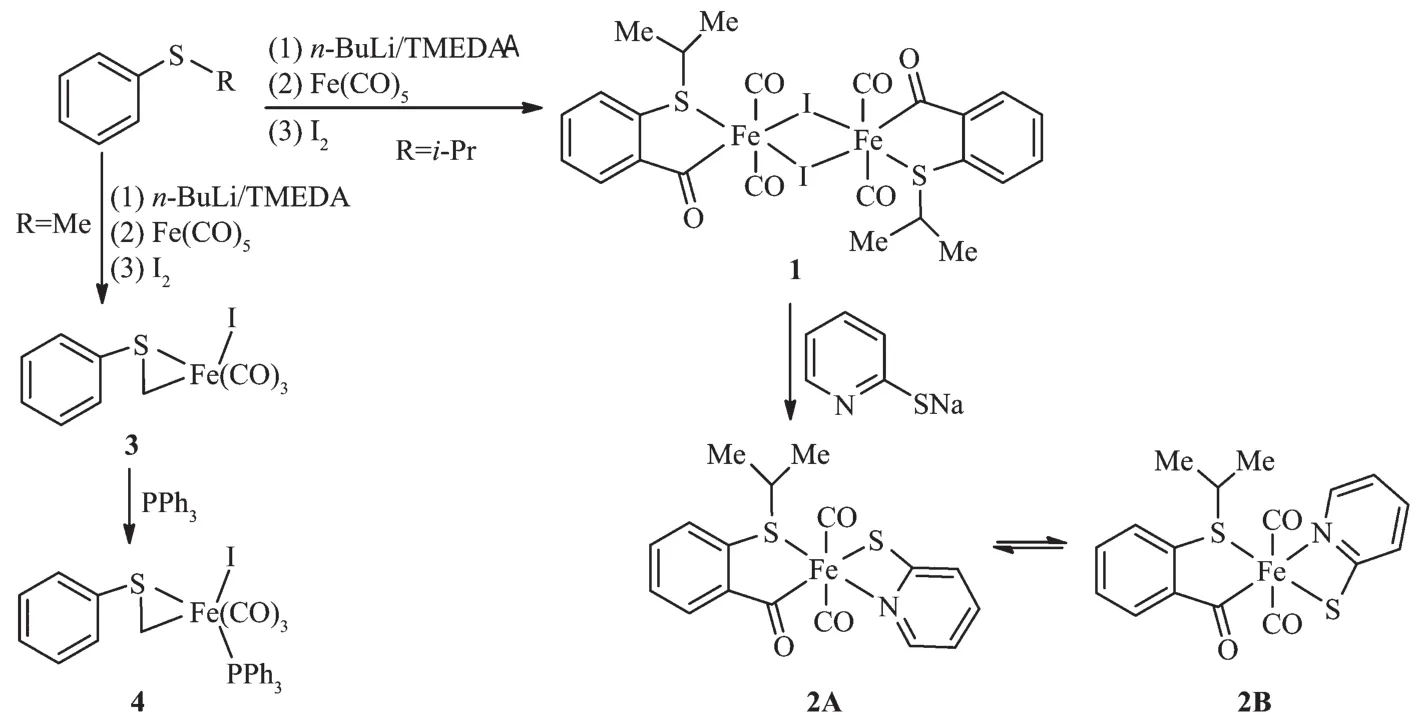
Scheme 1Synthesis and reactivity of thiobenzoyl and thiomethyl iron complexes
The IR spectra of 1 and 2 show that the acyl carbonyl peak occurrs at 1 628 and 1 615 cm-1,respectively,lower than those in polydentate acyl ligated iron complexes[22-23],but comparable to those in monodentate acyl ligated iron complexes[23].The corresponding acyl carbon resonates at 246.9 in the13C NMR spectrum of 1.The1H and13C NMR spectra of 2 contain two sets of proton and carbon signals,indicating that this complex possibly exists in solution as mixture of two geometric isomers(Scheme 1).The main structural difference of two isomers arises from the relative position of ortho-isopropylthiobenzoyl with 2-pyridinethiolate.Furthermore,the relative ratio of two isomers is variable in the CDCl3and acetone-d6solutions,showing that the interconversion between two isomers is possible in solution with the help of solvent,through partial dissociation of the ligands and succedent association process.Similar isomerism and interconversion have been observed in(pyrazol-1-yl) acyl iron complexes[23].
The structures of 1 and 2A were confirmed by X-ray crystallography,and are shown in Fig.1 and 2, respectively.The selected bond distances and angles are listed in Table 2.Fig.1 shows that complex 1 consists of two unequal iodide bridges between two benzoyl iron fragments.The Fe-I(1)bond distance (0.266 8(2)nm)is slightly shorter than the Fe-I(1A) bond distance(0.273 4(2)nm).These values are similar to those in bidentate acyl ligated iron complexes,but shorter than those in monodentate acyl ligated iron complexes[23].Fig.2 shows that the pyridyl nitrogen atom in 2A occupies the cis-position to the acyl group with the C(8)-Fe(1)-N(1)angle of 94.54(3)°.Complexes 1 and 2A possess analogous Fe-Cacyland C-Cacylbond distances,which are also comparable to the correspon-dingvaluesreportedformonodentateacyliron complexes[23].
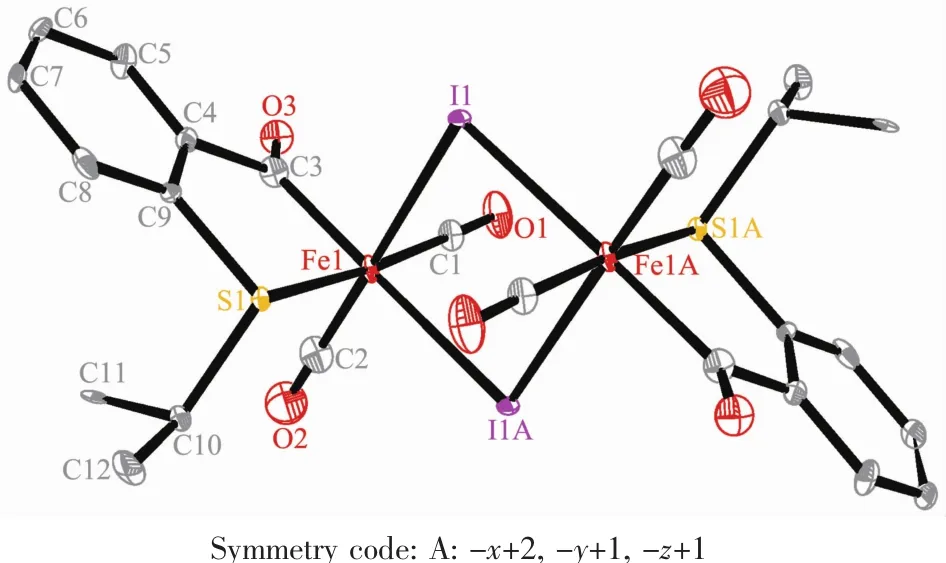
Fig.1 Molecular structure of 1 with 30%probability displacement ellipsoids

Fig.2 Molecular structure of 2A with 30%probability displacement ellipsoids

Table2 Selected bond distances(nm)and angles(°)for complexes 1,2A,3 and 4

Continued Table 1
2.2 Synthesis and reactivity of phenylthiomethyl iron complex
The lithiation process of thioanisole with n-BuLi is different from that of isopropylthiobenzene[30,32].Thioanisole was deprotonated with n-BuLi to give the methyl metalation product instead of the ortho lithiation product of phenyl group[32].Reactions of α-thiocarbanions with iron carbonyl have been investigated,which generated acyl iron intermediates and subsequently transferredtovariousorganicandorganometallic derivatives[33-34].However,we find that reaction of phenylthiomethyllithium with iron carbonyl followed by iodine only affords phenylthiomethyl iron complex 3(Scheme 1).No acyl iron derivatives are obtained. Reactivity of 3 is significantly different from that of 1. Forexample,similartreatmentof3with2-pyridinethiolate results in the decomposition of 3, rather than the substitution of iodide.In addition, reactionofalkylmetalcarbonylcompoundswith nucleophilic ligands,such as phosphine(PR3),has been extensivelyusedtoformacylmetalderivatives through alkyl/carbonyl migratory insertion reaction[9], but reaction of 3 with PPh3only give the carbonyl substitution product 4.No induced carbonyl insertion reaction took place,though such reaction should be able to reduce the ring strain in 3.Complexes 3 and 4 also are air-stable solids.Their IR and NMR spectra are highly consistent with the alkyl iron structure. These two complexes only exhibit three(for 3)or two (for 4)absorption bands in the range of 2 077~1 950 cm-1for terminal carbonyl ligands,and no acyl carbonyl absorption band is observed in their IR spectra.
Complexes 3 and 4 were also characterized by single crystal X-ray structural determination.Fig.3 and 4 reveal that the fundamental frameworks in complexes 3 and 4 are similar to each other,exceptthat PPh3locates on the trans-position of the methyl carbon in 4 instead of one carbonyl group like in 3. The phenylthiomethyl group bonds to the iron atom in a bidentate κ2-[C,S]chelating fashion in these two complexes,yielding a three-membered metallocyclic structure.Complexes 3 and 4 possess analogous Fe-S, Fe-I and Fe-Cmethylbond distances(Table 2).The Fe-S and Fe-I bond distances are also comparable to the corresponding values in 1 and 2.
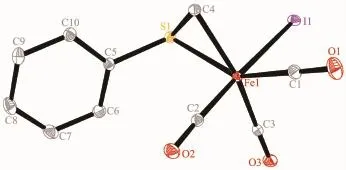
Fig.3 Molecular structure of 3 with 30%probability displacement ellipsoids

Fig.4 Molecular structure of 4 with 30%probability displacement ellipsoids
3 Conclusions
Insummary,ortho-isopropylthiobenzoyland phenylthiomethyl iron derivatives have been obtained by the reaction of ortho-isopropylthiophenyllithium or phenylthiomethyllithium with iron carbonyl followed by treatment with iodine.These complexes also show markedly different reactivities upon treatment with nucleophilic ligands.
[1]Almenara N,Ibarlucea L,Mendicute-Fierro C,et al.Dalton Trans.,2016,45:18502-18509
[2]Panthi B D,Gipson S L,Franken A.Inorg.Chim.Acta,2015, 425:176-181
[3]Wotal A C,Ribson R D,Weix D J.Organometallics,2014,33: 5874-5881
[4]Hermanson J R,Figley T M,Seibert A L,et al.J.Organomet. Chem.,2008,693:2061-2064
[5]Omae I.Coord.Chem.Rev.,2011,255:139-160
[6]Joe C L,Doyle A G.Angew.Chem.Int.Ed.,2016,55:4040-4043
[7]SU Lü(苏吕),XIAO Han-Bing(肖含兵),YUAN Yu-Meng(苑雨萌),et al.Chin.J.Org.Chem.(有机化学),2017,37:630-635
[8]CUI Long(崔龙),ZHOU Xi(周喜),YANG Xian-Gui(杨先贵), et al.Chinese J.Inorg.Chem.(无机化学学报),2014,30(7): 1600-1608
[9]Wang X,Cao K,Liu Y,et al.J.Am.Chem.Soc.,2013,135: 3399-3402
[10]Murshid N,Rahman M A,Wang X.J.Organomet.Chem., 2016,819:109-114
[11]ZHANG Tian-Yong(张天永),SHENG Liao(盛了),LI Bin(李彬),et al.Chem.Res.Appl.(化学研究与应用),2015,27 (11):1609-1618
[12]Corr M J,Murphy J A.Chem.Soc.Rev.,2011,40:2279-2292
[13]Wright J A,Turrell P J,Pickett C J.Organometallics,2010, 29:6146-6156
[14]Hiromoto T,Ataka K,Pilak O,et al.FEBS Lett.,2009,583: 585-590
[15]Hu B,Chen X,Gong D,et al.Organometallics,2016,35: 2993-2998
[16]Xu T,Yin C J M,Wodrich M D,et al.J.Am.Chem.Soc., 2016,138:3270-3273
[17]Turrell P J,Hill A D,Ibrahim S K,et al.Dalton Trans., 2013,42:8140-8146
[18]Royer A M,Salomone-Stagni M,Rauchfuss T B,et al.J. Am.Chem.Soc.,2010,132:16997-17003
[19]Turrell P J,Wright J A,Peck J M T,et al.Angew.Chem. Int.Ed.,2010,49:7508-7511
[20]Chen D,Scopelliti R,Hu X.J.Am.Chem.Soc.,2010,132: 928-929
[21]Royer A M,Rauchfuss T B,Gray D L.Organometallics, 2009,28:3618-3620
[22]Zhao D W,Liu X L,Zhang X Y,et al.J.Organomet.Chem., 2015,780:56-62
[23]Zhao D W,Xu Y,Guo Y W,et al.J.Organomet.Chem., 2015,791:303-310
[24]Yang X,Hall M B.J.Am.Chem.Soc.,2009,131:10901-10908
[25]Murray K A,Wodrich M D,Hu X,et al.Chem.Eur.J., 2015,21:3987-3996
[26]Song L C,Xu K K,Han X F,et al.Inorg.Chem.,2016,55: 1258-1269
[27]Durgaprasad G,Xie Z L,Rose M J.Inorg.Chem.,2016,55: 386-389
[28]CrystalStructure 3.7.0,Crystalclear 1.36:Crystal Structure Analysis Package,Rigaku and Rigaku/MSC,TX,2000.
[29]Sheldrick G M.Acta Crystallogr.Sect.A,2008,A64:112-114
[30]Horner L,Lawson A J,Simons G.Phosphorus Sulfur Silicon Relat.Elem.,1982,12(3):353-356
[31]Chen D,Scopelliti R,Hu X.Angew.Chem.Int.Ed.,2010, 49:7512-7515
[32]Seebach D,Gabriel J,Hässig R.Helv.Chim.Acta,1984,67: 1083-1099
[33]Alper H,Fabre J L.Organometallics,1982,1:1037-1040
[34]Lotz S,Dillen J L M,van Dyk M M.J.Organomet.Chem., 1989,371:371-382
Synthesis and Reactivity of ortho-Isopropylthiobenzoyl and Phenylthiomethyl Iron Derivatives
ZHAO Da-WeiXU YueSUN Bao-ChuanWANG Zhi-HongTANG Liang-Fu*
(State Key Laboratory of Elemento-Organic Chemistry,College of Chemistry,Nankai University,Tianjin 300071,China)
Reaction of isopropylthiobenzene with n-BuLi,and subsequently with iron carbonyl and iodine gave a dimeric ortho-isopropylthiobenzoyl iron derivative[(o-iPrS)C6H4COFe(CO)2I]2through two iodide bridges,while similar reaction of thioanisole only yielded mononuclear phenylthiomethyl iron complex C6H5SCH2Fe(CO)3I.These two complexes show significantly different reactivities upon treatment with various nucleophiles.For example, reaction of[(o-iPrS)C6H4COFe(CO)2I]2with sodium 2-pyridinethiolate(PySNa)give mononuclear complex(o-iPrS) C6H4COFe(CO)2(SPy),while reaction of C6H5SCH2Fe(CO)3I with PySNa results in the decomposition of the starting material.On the other hand,reaction of C6H5SCH2Fe(CO)3I with PPh3give complex C6H5SCH2Fe(CO)2(PPh3)I,but no characterizable product is obtained from the similar reaction of[(o-iPrS)C6H4COFe(CO)2I]2.All these newly synthesized compounds have been characterized by physico-chemical and spectroscopic methods,and their structures were unambiguously determined by X-ray crystallography.CCDC:1545834,1;1545835,2A;1545836,3; 1545837,4.
thioether;iron carbonyl;metal-acyl complex;pyridine;phosphine ligand
O614.81+1
A
1001-4861(2017)09-1547-08
10.11862/CJIC.2017.192
2017-06-22。收修改稿日期:2017-07-20。
国家自然科学基金(No.21372124)和天津市自然科学基金(No.14JCYBJC19700)资助项目。*
。E-mail:lftang@nankai.edu.cn;会员登记号:S060015703M。
猜你喜欢
山西化工(2023年10期)2023-11-15 08:47:42
中国酿造(2022年1期)2022-02-07 13:09:30
化工设计通讯(2016年8期)2016-12-03 02:10:42
百科知识(2016年18期)2016-10-28 00:20:12
广州化工(2016年8期)2016-09-01 09:49:49
当代化工研究(2016年9期)2016-03-20 16:22:14
四川师范大学学报(自然科学版)(2015年1期)2015-02-28 14:07:30
浙江化工(2014年9期)2014-08-15 00:50:54
无机化学学报(2014年8期)2014-02-28 17:32:47
无机化学学报(2014年3期)2014-02-28 17:30:57
