
金属玻璃的键态特征与塑性起源∗
2017-09-09 01:02:56袁晨晨
物理学报 2017年17期
袁晨晨
(东南大学材料科学与工程学院,南京 211189)
金属玻璃的键态特征与塑性起源∗
袁晨晨†
(东南大学材料科学与工程学院,南京 211189)
(2017年6月1日收到;2017年6月27日收到修改稿)
由于缺乏位错、晶界等典型的晶格缺陷,金属玻璃体系中承载力的形变单元为短程序或中程序原子团簇,键的强度及成键方向是影响原子间协调变形能力主要因素.本文通过与晶态合金对比,指出金属玻璃中原子键合方式与宏观力学性能的潜在关系,综述了金属材料电子结构与力学性能内在关系的最新研究进展,并系统介绍了金属玻璃电子结构特征、表征参量和主要测试手段,使读者对金属玻璃体系中原子间的键态特征有较清晰的认识,对进一步探索本征塑性较好的金属玻璃体系具有一定指导意义.
金属玻璃,电子结构,力学性能,塑性
1 引 言
长期以来,人们试图通过建立结构和性能的定量关系[1,2]来计算材料的性能并从理论上设计新材料.在晶态材料中,研究者们通过结构计算解释了低温下金属的形变机理[3],小原子半径非金属元素的晶界偏析现象[4−6]等.而在非晶态材料中,由于缺乏周期性的长程有序结构,至今很难建立结构和性能的定量关系.本文从电子结构角度出发介绍了近期国内外学者在建立微观电子结构和宏观塑性内在关系方面的研究工作,期望对探索新型大塑性金属玻璃合金体系有一定借鉴和启发作用.本文首先从晶态合金的电子结构和力学性能的复杂关系出发,介绍了金属玻璃的电子结构特征以及电子结构与力学性能的内在关系的研究进展,然后介绍了常用电子结构测试手段以及国内外学者在金属玻璃电子结构测试方面的最新研究成果.
2 电子结构与力学性能的内在关系
2.1 晶态材料中电子结构与力学性能的内在关系
关于合金体系的电子结构及其与力学性能相关性的研究已有很长的历史.材料学中的一些基本问题,如低温下bcc金属的位错滑移机理[3],B,C,P,S等元素的晶界偏析现象[5],均与微观电子结构密切相关.通常影响晶态金属材料力学行为的因素非常复杂,一般认为主要影响因素是微观尺度上的缺陷和晶体结构类型,但这无法解释具有相同晶格类型和晶格参数的纯金属和合金呈现出迥异的力学行为.例如密堆六方结构的纯金属Be在室温下很脆,在变形时更易于孪生而不是滑移[7,8],而同样结构的纯金属Ti拥有丰富的滑移带,表现出良好的延展性[9].显然,仅讨论原子排列及原子尺寸差异是不够的,必须考虑原子结合性质或电子结构起源(化学因素).理论计算表明,Ti的位错滑移能仅为Be的1/2左右[10],使得纯金属Ti在变形过程中比Be更容易开动位错,产生更多的滑移带.

图1 不同的B2结构金属间化合物RM{110}面内键的电荷分布[12] (a)不同稀土元素组成的Cu基B2化合物;(b)不同过渡族元素(从第八族到十一族)组成ScM相的键的电荷分布,这里将电荷分布图按照过渡族金属的d电子数目(从7到10依次增大)排列;稀土元素R在图片的中心,周围被过渡族金属M包围;红色代表成键后电子密度的增加,蓝色代表电荷密度的流失Fig.1.Bonding charge distribution(BCD)on B2{1 1 0}plane for various RMintermetallic compounds[12]:(a)The Cu-based B2 compounds with di ff erent earth elements;(b)the BCD panels arranged as the number of transition metal d electrons increase froM7 to 10(i.e.froMgroup 8 to group 11)in the ScMphases.The rare earth atoMis in the center of each plot and is surrounded by four Mmetal atoms.Red means an increase of the charge accumulated(i.e.electron density)after bonding,deep blue means a charge density loss.
当不同类型的原子形成合金时,原子间的电子轨道杂化导致了金属键的方向性.理论计算表明,合金中存在化学短程序(如TM-Al合金中TM偏好与Al最近邻,而非TM-TM/Al-Al最近邻,即异类原子间结合)[11],这种短程序在一定程度上影响着合金的塑性.Gschneidner等[12]的研究指出,由于原子间的成键方式不同,B2结构的金属间化合物塑性变形能力存在显著差别,如图1所示.Maclaren等[13,14]通过对晶体材料堆垛层错能的理论计算证实原子的置换将扰乱材料的化学短程序.Eberhart和Vvedensky[15]通过理论计算证明晶界电子结构的局域化特征降低了力学行为的稳定性,这是多晶体材料沿晶断裂的主要起因.早在1999年,Eberhart用金属键的方向性概念定量地解释了弹性剪切常数、弹性各向异性、同素异形体、合金结构以及为什么有些材料容易脆性断裂等诸多问题[16],通过系统的研究建立了电子结构和金属材料的强度、硬度、模量等力学参量的关系[17].在纯金属Al中也存在这种由电荷分布不均导致的方向性键,Nakashima等[18]通过会聚束电子衍射实验观察到了这一现象,并由此建立了弹性各向异性和原子结合性质或电子态密度分布之间的关系.金属的电荷密度分布能够反映金属键的方向性,在实验中能够通过电子结构测试的方法对其进行测量.合金材料的总电荷密度临界点(critical points of the total charge density)可以作为描述金属间化合物电子结构的参量,表征结构和力学性能的关系[19,20].Eberhart曾将化学反应的理论应用到现有的金属材料计算上来预测裂纹尖端的形变[21],应用这些化学概念所提供的定性信息描述裂纹尖端韧性和脆性行为的竞争机理——伴随着键的滑移和断裂,电荷的重新分布[22],并用黏结(cohesion and adhesion)概念解释bcc金属的沿晶或穿晶断裂以及受外界环境影响的脆化现象[23].2012年,Li等[24]通过第一性原理计算指出除了应变、沉淀、固溶等传统的基于位错运动的强化机理,在Al12W类型的金属间化合物中还存在由过饱和电子导致的共价强化机理,而化学键合的强弱可以通过引入“多余”电子(extra-electron)来调控.如图2,可以看到随着Al—Al,Al—X共价键的形成,弹性模量(G,E)显著增大.

图2 过饱和电子导致的共价强化(a)—(d),FCC-Al,Al12W类型Al12X(X=Re,□,He)的总电子态密度(□代表元素X被空穴替换);(e)和(f)分别代表Al12Re中Al—Al共价键所连接的最近邻二十面体电荷密度沿(020)面的截面图和Al12二十面体中Al—Re共价键沿(0y0)面的截面图;红色和蓝色等值线分别对应电荷的集中和分散程度;(g)Al12□,FCC-Al,Al12X(X=Cr,Mo,W,Mn,Tc,Re)体系计算得到的体模量(B)、杨氏模量(E)、剪切模量(G)、Cauchy pressure C12-C44、Pugh’s模量比(G/B)等各参量的比较[24]Fig.2.Extra-electron induced covalent strengthening:(a)–(d)Total DFT electronic densities of states of FCC-Al,Al12W-type Al12X(X=Re,□and He),□denotes that X is replaced by a vacancy;(e)and(f),section contour maps of the di ff erence of charge densities for(e)the Al—Al covalent bonds connecting the nearest-neighboring icosahedra as illustrated by the(020)plane and(f)the intra Al—Re covalent bonds within the Al12 icosahedron in the(0y0)plane of Al12Re,the red and blue isovalues correspond to the charge accumulations and depletions,respectively;(g)the comparison of calculated bulk moduli(B in GPa),Young moduli(E in GPa),shear moduli(G in GPa)and Cauchy pressure C12-C44 as well as Pugh’s modulus ratio of G/B(right side)in the series of Al12□,FCC Al,and Al12X(X=Cr,Mo,W,Mn,Tc and Re)[24].
2.2 金属玻璃的电子结构研究现状
在晶态材料中,力学性能主要依赖于原子的周期性排列方式以及它们之间的相互作用[25−27],结构和性能之间的内在关系已经被广泛地认识[3,15,22,28,29].然而,对于金属玻璃这类无序材料,关于结构和性能的关系尚未形成完备的理论体系.不过人们已经发现,除了长程无序,短程及中程序原子结构、金属玻璃的力学行为对组元元素的物理化学性质也有很强的依赖性[30−33].但是,在这种无序体系中寻找结构和性能的定量关系是一项非常艰巨的工作.由于缺乏位错、晶界等典型的晶格缺陷,研究认为金属玻璃体系中承载力的形变单元为几十至几百个原子组成的短程序或中程序原子团簇[34−37].在这样的一个个原子团簇中,协调变形的能力与键的长短及成键方向密切相关,也就是说金属玻璃的原子结合性质有可能成为影响力学性能的主要因素.研究表明,金属玻璃的强度、模量等力学参数与体系的密度、玻璃转变温度(Tg)、脆性指数(m)等物性参量密切相关[38,39],取决于组成元素的本征特性以及不同种类原子间键的结合强度和极化方向[40],因此金属玻璃成键方式的差异[30]可能对宏观性能产生直接影响.2007年,Fukuhara等[41]从电子结构的观点提出金属玻璃的合成准则,发现玻璃形成元素间的结合强度比晶态合金的理论预测值大很多,暗示spd和spf杂化键的存在,表明“共价键合”可能是提高金属玻璃形成能力的关键因素.大量的电子结构计算和实验均表明玻璃态和液态金属中金属和类金属原子间容易形成方向性的“共价键”[42−45],如Co-B[46],Fe-P-(B,C)[47],Zr-(Cu,Co,Ni)-Al[48],它们的存在可能会对金属玻璃的力学性能产生不良影响[48],但一直缺乏足够的实验证据.2009年,Ma等[49]在对金属玻璃体系电子结构和微观拓扑结构的第一原理计算中发现,类金属原子Al和过渡族元素Zr之间的pd轨道杂化可能是合金中存在方向性键的主要起因,导致Al-TM键缩短.第一原理计算表明,这一杂化会导致s电子在费米能级附近被d电子散射,即费米能级s电子态密度gs(EF)降低[50],而有方向性的d电子态密度的高低与金属玻璃的韧性和强度密切相关[48,51].2014年,Wang等[52]在与Eberhart的合作研究中,通过第一性原理计算指出金属玻璃脆性行为对键的性质如在临界点处电荷密度的分布状态很敏感.如图3,在Cu-Zr体系中用Al原子置换Cu原子将破坏Zr—Zr键,伴随着结构弛豫,Zr原子间的相对距离变大.Zr—Zr键的破坏使体系的亲电性大大降低,因此这种替换导致泊松比降低,表现为脆性断裂行为.可见,在金属玻璃体系中,合金元素间的原子键合作用即电子结构对体系的宏观塑性影响显著.虽然以上研究指出金属玻璃体系中的类金属元素如P,B,C,Al,与过渡族元素间易形成杂化的类共价键[42],显著恶化体系的力学性能[42,48,53],但是为了提高金属玻璃的形成能力,得到综合性能优异的大块金属玻璃体系,人们通常在合金体系内添加了较多的类金属元素,例如以铁族元素为基本组元的铁磁性金属玻璃体系Fe-Si-B[54],Fe-P-C[55],或者以贵金属元素为基本组元的顺磁性金属玻璃体系Pd-Ni-Cu-P[56],Cu-Zr-Al[57].然而该类金属玻璃体系的室温塑性变形能力都差,绝大多数大块金属玻璃体系的室温压缩塑性应变小,特别是具有大非晶形成能力的“非晶钢”在弹性变形阶段即发生灾难性断裂[53,58,59],极大地限制了金属玻璃作为功能和结构材料的工业应用前景.因此,对电子结构的系统研究对于深入理解电子结构和力学性能的内在关系,进一步改善金属玻璃体系的宏观塑性至关重要.
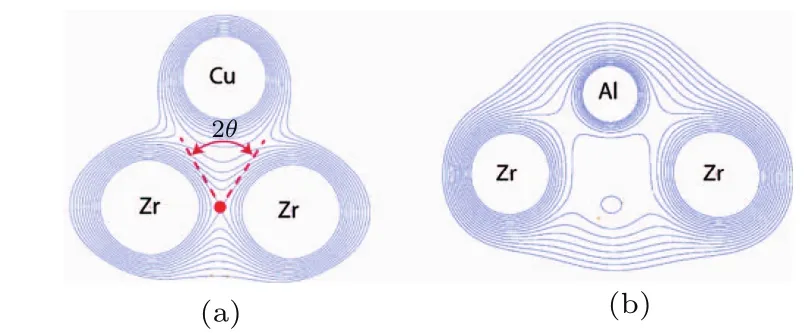
图3 Cu-Zr-Al的电荷密度截面图 (a)Cu-Zr环境的电荷密度截面图;(b)Al原子替换Cu原子的电荷密度截面图[52]Fig.3.Section contour maps of the charge density of Cu-Zr-Al:(a)Charge densities of all the CuZr environments;(b)charge densities resulting froMa substitution of a single Al atoMto a Cu site[52].
3 常用的电子结构测试手段
3.1 金属玻璃的电子结构特征
单电子近似是分析金属中电子结构和运动方式的理论基础.该理论假设各个电子的运动相互独立,每个电子在具有晶格周期性的势场V(r)中运动,V(r)为包括原子实和其他电子的平均势场.理想晶体的势场是周期性函数,哈密顿量具有晶格的平移对称性,存在平移对称量子数,称为简约波数k.通常用能量本征值En(k)来表示晶体的能带结构,其中E为k的函数.而在无序的金属玻璃体系中V(r)不是周期性函数,因而不存在好的量子数k和En(k),只能用基于单电子近似的能态密度函数g(E)来描述能带结构[60].金属玻璃虽然长程无序,但是对于一些原子间化学作用势较强的体系,仍然存在化学短程序或中程序结构[49,61,62],在外界条件如压力下表现出长程的拓扑序结构[63].实验证据和理论计算表明金属玻璃的局域结构及电子态与其晶体对应物(crystalline counterparts)相似[61,62,64],主要取决于元素间的化学作用及排列的紧密程度或相互作用距离,在计算中常用晶体化合物的g(E)对相近成分的金属玻璃电子态密度进行预测[64].
3.2 电子结构参量与表征手段
在晶体中,对电子结构的研究主要集中于构建费米面.费米面的形状和尺寸决定了电子输运、光学以及热平衡态性质.几种常用的测试费米面的手段,如回旋加速器谐振(cyclotron resonance),德哈斯-范.阿尔芬效应(de Haas-van Alphen e ff ect)通常要求在较低的温度下对纯度较高的单晶体进行测试,无序体系如金属玻璃成分均一,低温下没有特征相变,比较适合进行上面的研究[65].简单的碱土基金属玻璃如CaMgZn,CaAl体系的电子散射很弱,近似于自由电子状态,费米面仍然存在且接近球形,可以进行上述测试,但对于大多数金属玻璃体系,如典型的过渡族金属无序体系,电子散射很强,k不是一个好的量子数,费米面的概念并不存在,并不能用上面的方法进行电子结构的研究.基于单电子近似的能态密度函数g(E)并不依赖于平移波矢k,可以用来描述这类无序系统的电子结构[60].一些实验方法如紫外光电子能谱(UPS)和X射线光电子能谱(XPS)可以直接测试g(E),除此之外还有软X射线谱(SXS)、俄歇电子能谱(AES)[66−68].这些技术的原理相似,都是用近单色的电子或光子束聚焦样品,通过对反射的电子或光子进行能量分析获得体系的电子结构信息.但是这些方法都不能选择性地测量单个组分元素的分波电子态密度的贡献.以Zr70Cu30金属玻璃为例,它的UPS曲线由Zr的4d,5p,5s和Cu的3d,4p,4s电子态的多重轨道作用贡献[67,68].此外,UPS测试获得费米能级附近的强度值也不能够直接对应费米能级附近的电子态密度g(EF),因为不同的轨道贡献是通过跃迁概率加权平均得到,最终的态密度取决于电子的跃迁概率[69].但是对于不同的测试手段,d,p和s电子态对g(EF)的贡献不同,通过结合XPS,UPS和AES等测试手段可以大致区分各个分波电子态的作用.
除了这些实验测试手段之外,也可以通过测试一些物性参量如比热、磁化率、核磁共振(NMR)金属位移等计算得到费米能级附近的电子态密度g(EF).这里对几个常用参数进行详细介绍,它们均是g(EF)的函数,其中能够直接反映电子态密度的是材料的电子比热Cel,可表示为
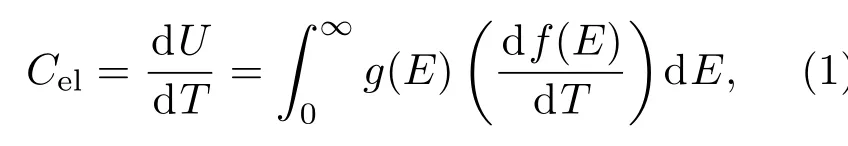

(2)式成立的重要前提是g(E)在接近EF时不发生突变,大多数金属玻璃体系均满足上述要求.但也有电子比热项不能从总比热C中单独分离出来的情况,如铁磁性金属玻璃体系Fe20Ni60P14B6在低温下仍然有磁的贡献,难以区分电子部分的贡献[70].当然,为了获得g(EF)的精确值,还必须考虑电子-声子重整化作用(electron-phonon renormalization)[71].
反映g(EF)的第二个主要参数是磁化率,在自由电子近似中,泡利顺磁磁化率χP可表示为

其中,µB为玻尔磁子.这一关系只对自由电子体系成立,通常实验得到的磁化率为总磁化率χ,主要包括泡利顺磁(χP)和轨道(χvv和χd)贡献,后者在过渡族元素中占主导地位,不可忽略.因此需要参照其他的物理参量来获得更清晰的电子结构图像,而最常用的参数是核磁共振各向同性金属位移Kiso,可表示为
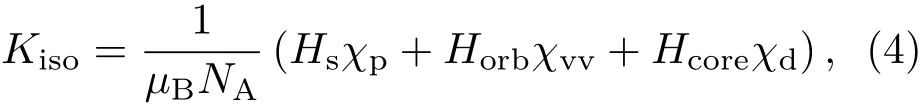
其中Hs,Horb和Hcore为不同电子作用在原子核位置的超精细场,NA是阿伏伽德罗常数.通过分析金属位移和磁化率参量间的关系可以获得不同超精细场的作用[72,73].此外,核磁共振技术本身也是研究金属材料电子结构的重要手段之一,除了金属位移我们还可以获得核周围的电场梯度等物理参量.由核磁共振频谱的线形分析,我们可以获得很多结构方面的信息[74],比如四极矩、偶极矩这些参量能够给出金属玻璃局域原子结构对称性[75,76]、短程序[77−79]、中程序[80]、原子间距[81]等结构信息.理论上NMR可以给出任一组元原子核周围的电子结构信息以及不同分波电子态的贡献,比如说在ZrTMAl金属玻璃体系中通过金属位移随成分的变化可以将Al核周围d电子的作用和其他电子超精细作用区分出来[51],这对于建立清晰的结构和性能关系至关重要.
近期我们通过27Al[82]和93Nb[83]核磁共振谱研究了含Al,Nb元素的金属玻璃及金属间化合物的局域电子结构或结合方式、韧脆转变过程中模量和电子结构的变化及其与力学性能的关系.由图4可以看到,随着Sn含量的变化,Zr61Cu18.3−xNi12.8Al7.9Snx块体金属玻璃体系(0≤x≤2.5)的断裂能和27Al金属位移具有相似的变化趋势,也就是说金属玻璃的塑性变形能力与核磁共振谱揭示的费米能级s电子态密度gs(EF)的高低相关联,直接反映了体系内原子尺度上裂纹尖端能量耗散的物理机理[82].

图4 Zr61Cu18.3−xNi12.8Al7.9Snx块体金属玻璃体系(0≤ x≤ 2.5)的27Al核磁共振金属位移(左侧坐标轴)和断裂能(右侧坐标轴)随Sn含量的变化[82]Fig.4.The correlation of27Al NMR line shift(left coordinate)and fracture energy(right coordinate)with Sn concentration[82].
2011年,He等[48]报道了Zr(Cu/Ni)Al金属玻璃和化合物的电子结构的第一原理计算的结果,发现将Ni替换Cu,Ni将与Al的sp轨道发生强烈的杂化作用而降低合金材料的断裂韧性,表现为Ni的半填满d电子态更接近费米能级.对于所有3p和4d TM-Al化合物稳态相,电子结构计算显示Al原子的p轨道和TM原子的d轨道在能量空间上重叠,表现出由杂化导致的方向性键,见图5[84].虽然由于费米能级附近有较高的d电子态,这类化合物仍然具有较好的金属性.Al原子和过渡族金属原子间强烈的pd杂化是导致Al-Al原子sp能带劈裂以及Al元素的3s电子态密度在费米能级附近较低的主要原因,这里Al元素的3s和TM元素的3d电子态密度的变化可以分别由金属位移和磁化率反映[84].Al原子的3s电子跃迁到费米能级以下的较低能态,该能态接近基态能级,是非成键态.在我们研究的Zr-Cu-Ni-Al-Sn体系中Sn原子的添加使Al原子周围的3s电子态进一步减小,从而增加了Al原子周围金属键的方向性.由于pd杂化原子轨道的范围仅在一个原子距离内,而s轨道的范围超出两个最近邻原子,拥有更大的重叠度,在剪切过程中类s原子轨道重叠度基本保持不变,而pd杂化原子轨道容易剪切超过某一平衡位置使得键的轨道重叠度减少到远低于类s轨道,使结合键断开,表现出较差的宏观本征塑性,如图6所示.该项研究指出gs(EF)可以作为结构序参量评估金属玻璃的本征塑性.Shen等[85]在Fe基金属玻璃体系Fe-Ni-P-C中也发现了类似的Fe族元素的3d与非金属元素P和C电子态的pd杂化现象,他们利用XPS监测到Fe80−xNixP13C7(0≤x≤30)金属玻璃体系中,费米能级上总的电子态密度随Ni含量的变化而变化的现象.该研究表明费米能级附近总的电子态密度变化是由电子轨道杂化而导致的s电子态密度的散射造成的,即高的s电子态密度与弱的pd杂化相对应.而gs(EF)越高,原子团簇内键的柔顺度则越高,使金属玻璃体系中存在均匀分布的剪切带,表现出较好的塑性变形能力.
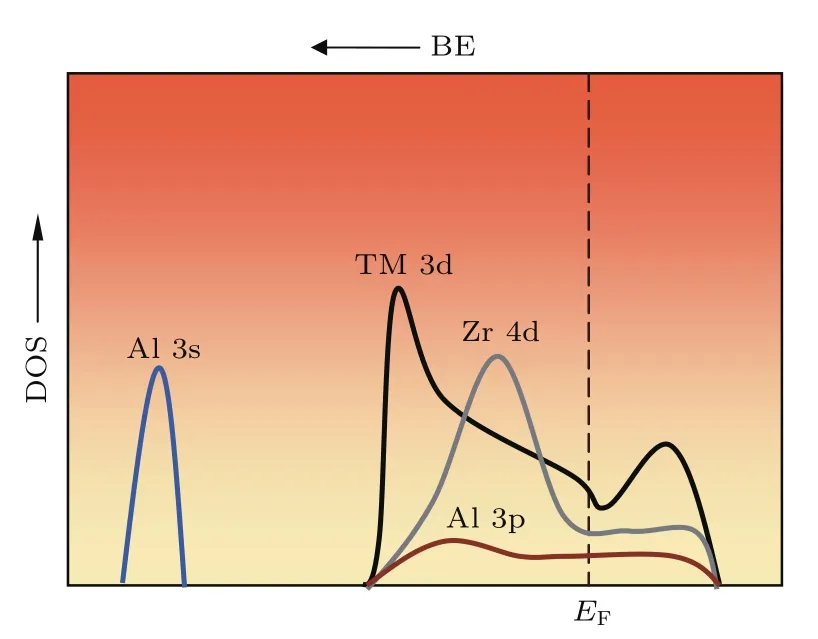
图5 电子态密度分布示意图,仅显示了Al 3s,Al 3p和过渡族金属TM(Co,Ni,Cu)3d和Zr 4d态[84]Fig.5.A schematic diagraMof density of states(DOS)shape against binding energy(BE).For clarity,only Al 3s,Al 3p and TM(Co,Ni,Cu)3d and Zr 4d states are illustrated[84].
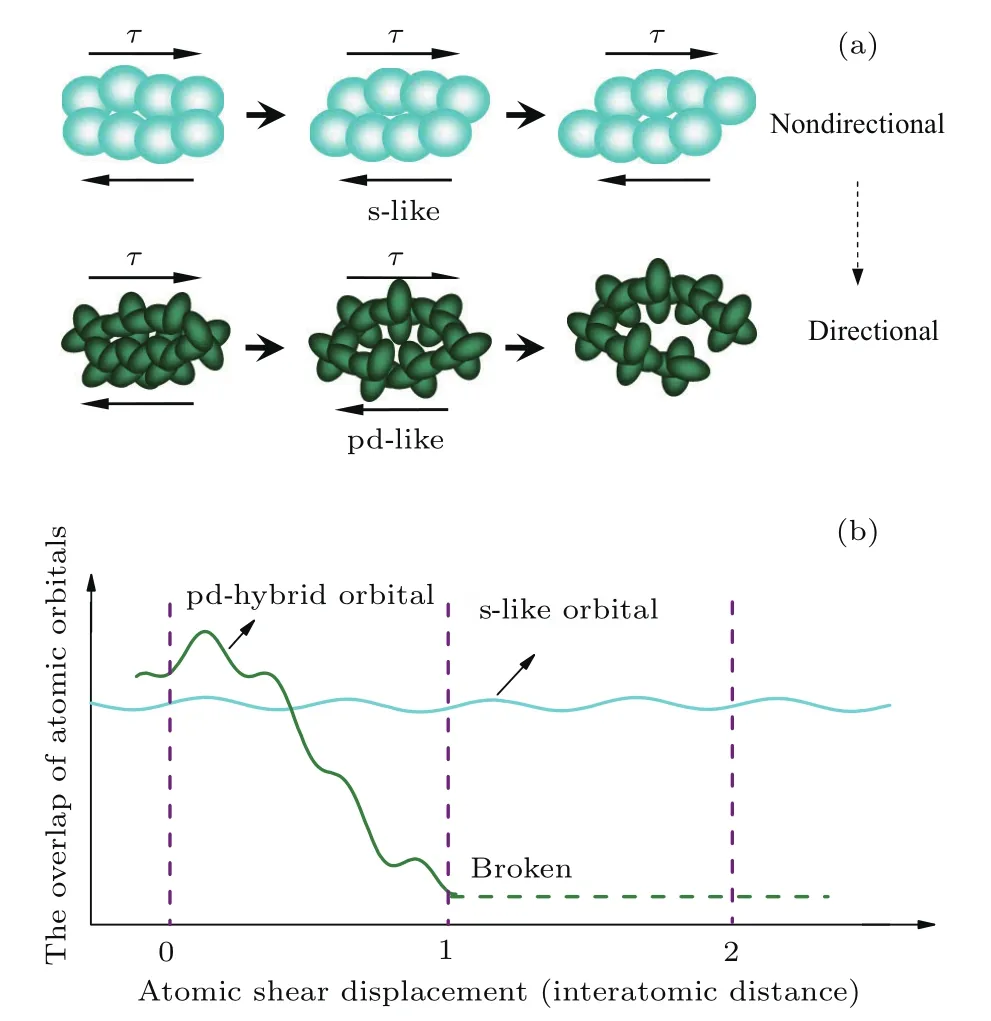
图6 (a)剪切过程中非极化类s和pd杂化原子轨道重叠程度示意图;(b)原子轨道的重叠度和原子剪切位移关系图[82]Fig.6.(a)A schematic illustration of the overlap of nonpolarizable s-like and pd hybrid atomic orbitals during shear;(b)the overlap of atomic orbital versus the atomic shear displacement[82].
4 结 论
在金属玻璃体系中,短程序或中程序原子团簇是承载力的基本单元,团簇内部原子间协调变形的能力与原子结合强度及成键方向密切相关.而费米能级附近的电子态密度g(EF)的变化则直接反映了材料中方向性键的形成.费米能级附近s电子态密度gs(EF)越高(原子键的杂化程度越低)则键的柔顺度越高,表现为本征塑性越好.实验上我们可以通过改变成分配比等内在因素调节合金材料gs(EF)的大小来改善金属玻璃的本征脆性,甚至还可以通过改变体系的环境压力、温度等外在因素适当地调控费米能级附近的电子态密度g(EF).在计算机模拟中,我们用晶体化合物的电子态密度g(E)对相近成分的金属玻璃电子态密度进行预测.除此之外,还可以利用XPS,UPS和AES等实验测试手段结合一些物理参数如比热、磁化率、NMR金属位移获取金属玻璃体系费米能级附近的电子态密度g(EF).然而,由于测试手段的限制,常用的电子结构和物性测试手段如XPS,UPS和AES等只能给出各元素总的电子态密度贡献,这使得我们很难明确建立微观电子结构和宏观力学性能的关系,这就需要研究者们相互合作,探究更有效的结构测试手段.
感谢中国科学院物理研究所汪卫华研究员的讨论以及东南大学侯龙硕士在文献资料整理方面提供的帮助.
[1]Eberhart ME,Clougherty D P 2004 Nat.Mater.3 659
[2]Eberhart ME,Giamei A F 1998 Mater.Sci.Eng.A:Struct.Mater.Prop.Microstruct.Process.248 287
[3]Jones T E,Eberhart ME,Clougherty D P,Woodward C 2008 Phys.Rev.Lett.101 085505.
[4]Liu Y,Chen K Y,Lu G,Zhang J H,Hu Z Q 1997 Acta Mater.45 1837
[5]Krasko G L,Olson G B 1990 Solid State Commun.76 247
[6]Lejcek P 2010 Grain Boundary Segregation in Metals(Vol.136)(Berlin,Heidelberg:Springer-Verlag Press)p1
[7]Sloman H A 1932 J.I.Met.49 365
[8]Lee H T,Brick R M1952 J.I.Met.4 147
[9]Rosi F D,Dube C A,Alexander B H 1953 Trans.Am.I.Min.Metall.Eng.197 257
[10]Pugh S F 1954 Philos.Mag.45 823
[11]Bader R F W 1998 J.Phys.Chem.A 102 7314
[12]Gschneidner K A,Ji M,Wang C Z,Ho K M,Russell A M,Mudryk Y,Becker A T,Larson J L 2009 Acta Mater.57 5876
[13]Maclaren J M,Crampin S,Vvedensky D D,Eberhart ME 1989 Phys.Rev.Lett.63 2586
[14]Maclaren J M,Gonis A,Schadler G 1992 Phys.Rev.B 45 14392.
[15]Eberhart ME,Vvedensky D D 1987 Phys.Rev.Lett.58 61
[16]Eberhart ME 1999 Sci.Am.281 66
[17]Eberhart ME 1996 Acta Mater.44 2495
[18]Nakashima P N H,Smith A E,Etheridge J,Muddle B C 2011 Science 331 1583
[19]Eberhart ME,Clougherty D P,Maclaren J M1993 Philos.Mag.B:Phys.Condens.Matter Stat.Mech.Electron.Opt.Magn.Prop.68 455
[20]Eberhart ME,Jones T E,Sauer MA 2008 JOM60 67
[21]Eberhart ME 1996 Philos.Mag.A:Phys.Condens.Matter Struct.Defect Mech.Prop.73 47
[22]Beltz G E,Selinger R L B,KiMK S,Marder MP 1999 Fracture and Ductile Vs.Brittle Behavior-Theory,Modelling and Experiment(Vol.539)(Cambridge:Cambridge University Press)p13
[23]EberhartME,Donovan,MM,Maclaren,JM,Clougherty,D P 1991 Prog.Surf.Sci.36 1
[24]Niu H Y,Chen X Q,Liu P T,Xing W W,ChengX Y,Li D Z,Li Y Y 2012 Sci.Rep.2 718
[25]Ogata S,Li J,Yip S 2002 Science 298 807
[26]Morinaga M,Saito J,Yukawa N,Adachi H 1990 Acta Metall.Mater.38 25
[27]Heredia F E,Pope D P 1991 Acta Metall.Mater.39 2017
[28]Lejcek P,Hofmann S 1995 Crit.Rev.Solid State Mater.Sci.20 1
[29]Datta A,Waghmare U V,Ramamurty U 2008 Acta Mater.56 2531
[30]Cheng Y Q,Cao A J,Ma E 2009 Acta Mater.57 3253
[31]Weaire D,Ashby MF,Logan J,Weins MJ 1971 Acta Metall.19 779
[32]Cheng Y Q,Ma E 2011 Acta Mater.59 1800
[33]Wang W H 2007 Prog.Mater.Sci.52 540
[34]Spaepen F 1977 Acta Metall.25 407
[35]Argon A S 1979 Acta Metall.27 47
[36]Spaepen F 2006 Scr.Mater.54 363
[37]Johnson W L,Samwer K 2005 Phys.Rev.Lett.95 195501
[38]Wang W H 2005 J.Non-Cryst.Solids 351 1481
[39]Wang W H 2006 J.Appl.Phys.99 093506
[40]Rouxel T 2007 J.Am.Ceram.Soc.90 3019
[41]Fukuhara M,Takahashi M,Kawazoe Y,Inoue A 2007 Appl.Phys.Lett.90 073114
[42]Mayou D,Nguyenmanh D,Pasturel A,Cyrotlackmann F 1986 Phys.Rev.B 33 3384
[43]Tamura R,Takeuchi T,Aoki C,Takeuchi S,Kiss T,Yokoya T,Shin S 2004 Phys.Rev.Lett.92 146402
[44]Huang L,Wang C Z,Hao S G,Kramer MJ,Ho K M2010 Phys.Rev.B 81 014108
[45]Yuan C C,Yang F,Kargl F,Holland-Moritz D,Simeoni G G,Meyer A 2015 Phys.Rev.B 91 214203
[46]Amamou A,Krill G 1979 Solid State Commun.31 971
[47]Amamou A 1979 Phys.Status Solidi A:Appl.Res.54 565
[48]He Q A,Cheng Y Q,Ma E,Xu J A 2011 Acta Mater.59 202
[49]Cheng Y Q,Ma E,Sheng H W 2009 Phys.Rev.Lett.102 245501
[50]Weinert M,Watson R E 1998 Phys.Rev.B 58 9732
[51]Sandor MT,Kecskes L J,He Q,Xu J,Wu Y 2011 Chin.Sci.Bull.56 3937
[52]Wang X F,Jones T E,Wu Y,Lu Z P,Halas S,DurakiewiczT,Eberhart ME 2014 J.Chem.Phys.141 024503
[53]Gu X J,Poon S J,Shi fl et G J,WidoMM2008 Acta Mater.56 88
[54]Mitra A,Jiles D C 1997 J.Magn.Magn.Mater.168 169
[55]Duwez P,Lin S C H 1967 J.Appl.Phys.38 4096
[56]Nishiyama N,Inoue A 1996 Mater.T.JIM37 1531
[57]Xu D H,Duan G,Johnson W L 2004 Phys.Rev.Lett.92 245504
[58]Shen J,Liang W Z,Sun J F 2006 Appl.Phys.Lett.89 121908
[59]Inoue A,Shen B L,Yavari A R,Greer A L 2003 J.Mater.Res.18 1487
[60]Mott N F,Davis E A 1979 Electronic Processes in Non-Crystalline Materials(Oxford:Clarendon Press)Chaps.5
[61]Miracle D B 2004 Nat.Mater.3 697
[62]Sheng H W,Luo W K,Alamgir F M,Bai J M,Ma E 2006 Nature 439 419
[63]Zeng Q,Sheng H,Ding Y,Wang L,Yang W,Jiang J Z,Mao W L,Mao H K 2011 Science 332 1404
[64]Xi X K,Sandor MT,Wang H J,Wang J Q,Hwang W,Wu Y 2011 J.Phys.-Condes.Matter 23 115501
[65]Johnson K H,Leon F A,Eberhart ME 1982 J.I.Met.34 58
[66]Oelhafen P,Hauser E,Guntherodt H J 1980 Helvet.Phys.Acta 52 378
[67]Oelhafen P,Hauser E,Guntherodt H J,Bennemann K H 1979 Phys.Rev.Lett.43 1134
[68]Amamou A 1980 Solid State Commun.33 1029
[69]Park R L 1975 Phys.Today 28 52
[70]Onn D G,Johnson W D,Gleeson P F,Donnelly T A,Egami T,Liebermann H H 1977 J.Phys.C:Solid State Phys.10 639
[71]Butler N H 1977 Phys.Rev.B:Solid State 15 5267
[72]Yang D P,Hines W A,Tsai C L,Giessen B C,Lu F C 1991 J.Appl.Phys.69 6225
[73]Hines W A,Glover K,Clark W G,Kabaco ffL T,Modzelewski C U,Hasegawa R,Duwez P 1980 Phys.Rev.B 21 3771
[74]Narath A 1969 Phys.Rev.179 359
[75]Panissod P,Guerra D A,Amamou A,Durand J,Johnson W L,Carter W L,Poon S J 1980 Phys.Rev.Lett.44 1465
[76]Xi X K,Li L L,Zhang B,Wang W H,Wu Y 2007 Phys.Rev.Lett.99 095501.
[77]Zhang Y D,Budnick J I,Ford J C,Hines W A 1991 J.Magn.Magn.Mater.100 13
[78]Pokatillov V S 2007 Phys.Solid State 49 2217
[79]Imafuku M,Saito K,Kanehashi K,Saida J,Sato S,Inoue A 2005 J.Non-Cryst.Solids 351 3587
[80]Breitzke H,Luders K,Scudino S,Kuhn U,Eckert J 2004 Phys.Rev.B 70 014201
[81]Panissod P,Bakonyi I,Hasegawa R 1983 Phys.Rev.B 28 2374
[82]Yuan C C,Xiang J F,Xi X K,Wang W H 2011 Phys.Rev.Lett.107 236403
[83]Yuan C C,Yang Y F,Xi X K 2013 J.Appl.Phys.114 213511
[84]Yuan C C,Shen X,Cui J,Gu L,Yu R C,Xi X K 2012 Appl.Phys.Lett.101 021902
[85]Yang W M,Liu H S,Zhao Y C,Inoue A,Jiang K M,Huo J T,Ling H B,Li Q,Shen B L 2014 Sci.Rep.4 6233
PACS:64.70.pe,71.23.Cq,87.15.La,62.20.fkDOI:10.7498/aps.66.176402
*Project supported by the National Natural Science Foundation of China(Grant Nos.51601038,51631003),the Natural Science Foundation of Jiangsu Province,China(Grant No.BK20171354),the National Basic Research PrograMof China(Grant No.2016YFB0300502),and the Fundamental Research Funds for the Central Universities,China(Grant No.2242017K40189).
†Corresponding author.E-mail:yuanchenchenneu@163.com
Bonding nature and the origin of ductility of metallic glasses∗
Yuan Chen-Chen†
(School of Materials Science and Engineering,Southeast University,Nanjing 211189,China)
1 June 2017;revised manuscript
27 June 2017)
Understanding the structure-property relationship of metallic glasses(MGs)at an atomic-or electronic level is a challenging topic in condensed matter physics.MGs usually exhibit low macroscopic plasticity,owing to the localized plastic fl ow in nano-and micro-meter scale shear bands upon deformation,which impedes their wide application as new structural materials.Thus,a detailed description of internal structure and establishing the structure-property relationship would underpin our knowledge of the mechanisms for the ductility/brittleness of MGs and further improve their plasticity.Due to the lack of structural defects such as dislocations and grain boundaries,the short-or middle-ranged ordered clusters are the typical deformation units in MGs,where the bonding strength and direction between atoms are the key factors that a ff ect the cooperative displacements inside deformation unit.However,the bonding nature of MGs and their structure-property relationship are little studied systematically,which hinders our comprehensive understanding the basic problems about mechanical behaviors of MGs,such as fracture and plasticity deformation mechanism.
In this paper,the potential correlation between the fl exibility of bonding and ductility of MGs is discussed in detail.The fi rst section gives a simple introduction of this topic.In the second section,the latest research progress of the electronic structural study of MGs is presented.Here,the corresponding studies of electronic structures of crystal alloys and their relationship with the mechanical properties are also presented for comparison.In the third section,the traditional and new experimental techniques employed for electronic structure measurements are presented,such as X-ray photoelectron spectroscopy,ultraviolet photoemission spectroscopy and auger electron spectroscopy and the parameters such as nuclear magnetic resonance knight shift,susceptibility(χ)and speci fi c heat(C)are also given in order to introduce electronic structure analysis methods of MGs and further reveal the bonding character of MGs and recent experimental fi ndings of the relationship between the electronic structure and the mechanical properties of MGs.
Numerous studies show that in the typical transition metal(TM)—metalloid metallic glass systems,the bond fl exibility or mobility of atoms at the tip of crack that depends on the degree of bonding hybridization,determines the intrinsic plasticity versus brittleness.For instance,in these transition metal(TM)-based MGs,when metalloid element Mwith sp-element shells is alloyed in the TMmatrix,the s-density of states(DOS)at Msites is scattered far below the Fermi level due to the pd hybridization between the p orbitals of Melement and the d orbitals of TM.This causes the reduction of s-DOS at the Fermi energy(gs(EF))at the solute Msites and exhibits a strong directional boningcharacter.Thus,it is proposed that the gs(EF)can be employed as an e ff ective order parameter to characterize the nature of bonding,especially in the aspect of evaluating bond fl exibilities in amorphous alloys.This shows that the plastic fl ow and fracture process of MGs on an atomic scale can be well described using a simple bonding model where the deformation process is accompanied with the broken-down and reforming of atomic bonding inside short-or middleranged ordered clusters,since the defects are absent in MGs.We hope that this introduction can provide a much clearer picture of the bonding character of MGs,and further guide us in understanding the mechanisMfor ductile-to-brittle transition in MGs and exploring the novel MGs with intrinsic plasticity.
metallic glasses,electronic structure,mechanical properties,ductility
10.7498/aps.66.176402
∗国家自然科学基金(批准号:51601038,51631003)、江苏省自然科学基金(批准号:BK20171354)、国家重点基础研究发展计划(批准号:2016YFB0300502)和中央高校基本科研业务费(批准号:2242017K40189)资助的课题.
†通信作者.E-mail:yuanchenchenneu@163.com
©2017中国物理学会Chinese Physical Society
http://wulixb.iphy.ac.cn
猜你喜欢
云南化工(2021年11期)2022-01-12 06:06:14
数学物理学报(2019年6期)2020-01-13 06:08:24
发明与创新·小学生(2019年12期)2019-12-05 06:02:28
发明与创新(2019年47期)2019-11-21 01:16:18
山东冶金(2019年3期)2019-07-10 00:54:00
陕西中医(2018年6期)2018-08-29 00:43:34
中国塑料(2016年1期)2016-05-17 06:13:00
读写算·教研版(2016年8期)2016-05-07 11:52:08
中国塑料(2016年11期)2016-04-16 05:25:55
焊接(2015年9期)2015-07-18 11:03:53
