
7-溴-2-庚酮的合成
2017-09-04 02:53:09杜远新罗树常
山东化工 2017年8期
杜远新,罗树常
(贵州工程应用技术学院 化学工程学院,贵州 毕节 551700)
7-溴-2-庚酮的合成
杜远新,罗树常
(贵州工程应用技术学院 化学工程学院,贵州 毕节 551700)
以6-溴己酸与乙二酰二氯为原料,经酰化氯代、Weinreb酰胺转化及格式试剂取代反应,以78.6%的总收率合成了7-溴-2-庚酮。中间体及产物结构均经过核磁氢谱和质谱进行表征。该路线具有工艺简单、操作方便及总收率高的优点,可适用于批量生产。
7-溴-2-庚酮;6-溴己酸;改进合成
7-溴-2-庚酮是一种重要的医药中间体,可用于合成多种抗肿瘤药物及周围血管扩张药己酮可可碱等[1-3]。目前,有很多小组报道了不同的合成路线[4-8],本文以6-溴己酸与乙二酰二氯为原料,经酰化氯代、Weinreb酰胺转化及格式试剂取代反应,以78.6%的总收率合成了7-溴-2-庚酮,合成路线如图1所示。中间体及产物结构均经过核磁氢谱和质谱进行表征。该路线具有工艺简单、操作方便及总收率高的优点,可适用于批量生产。
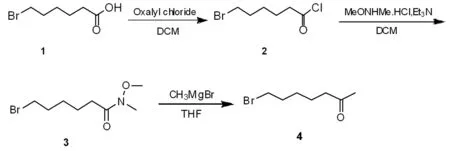
图1 7-溴-2-庚酮合成路线
Fig.1 The synthesizing route of 7-bromoheptan-2-one
1.1 6-溴己酰氯的合成
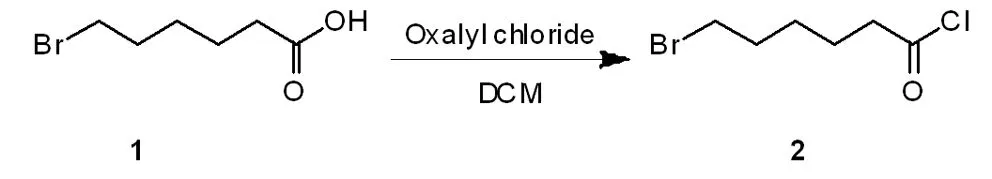
在氮气保护下,于0 ℃下用200 mL无水二氯甲烷溶解6-溴己酸20 g(103 mmol)在250 mL的圆底烧瓶中。滴加乙二酰二氯19.52 g (154 mmol)搅拌30 min。然后升温至25 ℃,搅拌3 h。薄层层析法(SiO2,比移值= 0.7、乙酸乙酯/石油醚= 3:1)(在反应的样本中添加甲醇,使其转化为甲酸酯)显示,起始原料消耗完成。旋转蒸发仪减压真空法下得到化合物2(6-溴己酰氯)约22 g(99 mmol),产率96%,其性状为黄色油状。没有进一步纯化用于下一步。
1.2 注意事项
生成的6-溴己酰氯不能与醇类化合物接触,其化合物的氯元素极易被醇取代从而生成酸酯。
2 6-溴己酰氯生成6-溴-N-甲氧基-N-甲基酰胺
2.1 合成路线及步骤
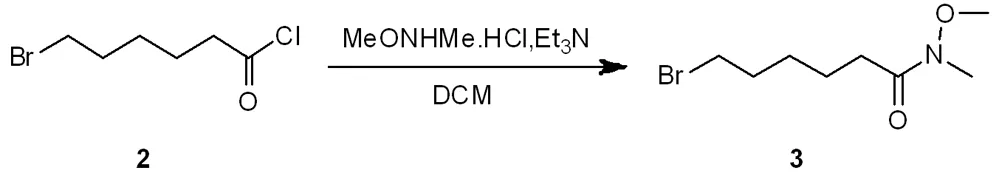
在氮气保护下,将盐酸N,O-二甲基羟胺12.06 g (124 mmol)和三乙胺28.7 mL(206 mmol)称量在250 mL圆底烧瓶中,用150 mL无水二氯甲烷将其溶解,于0 ℃下滴加6-溴己酰氯22 g(103 mmol),搅拌30 min,升温至25 ℃搅拌18 h。LC-MS (MS (ESI) m/z: 240.0 [M + H+], tR= 1.017 min。用50 mL的无水二氯甲烷稀释。有机相加入50 mL的水清洗 ,20 mL 1 mol/L 盐酸溶液中和有机相中的碱,用80 mL的二氯甲烷萃取三次,100 mL的饱和氯化钠水洗。有机相加入无水硫酸钠干燥,旋转蒸发减压真空下得到化合物3(6-溴-N-甲氧基-N-甲基酰胺) 23.2 g(93 mmol),收率90%。性状为黄色的油状物。进行核磁氢谱和质谱确证。
2.2 质谱及核磁谱图
质谱及核磁谱图见图2。
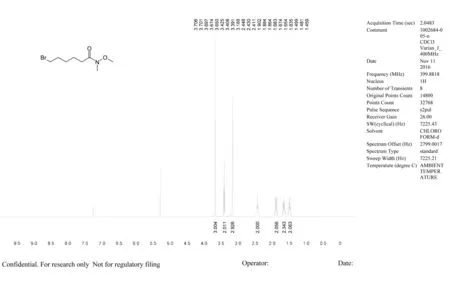
ESI-MS m/z [M + H+]:240.0, tR= 1.017 min
1H NMR (400 MHz, CHLOROFORM-d) δ : 1.40 - 1.54 (m, 2 H) 1.57~1.73 (m, 2 H) 1.8~ 1.98 (m,2 H) 2.43 (br t, J=7.39 Hz, 2 H) 3.17 (s, 3 H) 3.41 (t, J=6.73 Hz, 2 H) 3.63 - 3.73 (m, 3 H)
图2 质谱及核磁谱图
3 6-溴-N-甲氧基-N-甲基酰胺合成7-溴-2-庚酮
3.1 合成路线及操作步骤
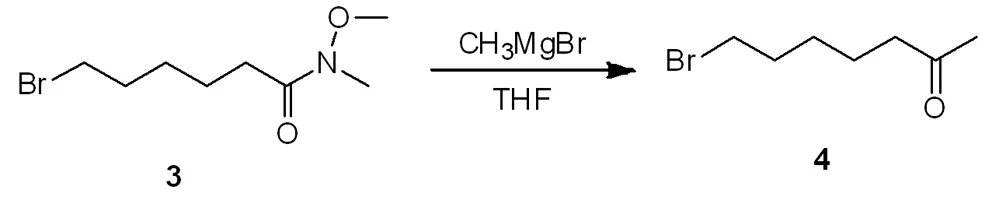
氮气保护下,将6-溴-N-甲氧基-N-甲基酰胺22 g (92 mmol)于清洗干燥好的圆底烧瓶中,加入200 mL无水四氢呋喃,于0 ℃下滴加甲基溴化镁61.6 mL(185 mmol),冰盐浴搅拌反应3.5 h。薄层层析法(SiO2,比移值= 0.31、乙酸乙酯/石油醚= 5:1)显示,起始原料反应完后用1 mol/L 的盐酸溶液50 mL淬灭。加入100 mL水洗,用150 mL的乙酸乙酯萃取三次。再用100 mL饱和氯化钠溶液进行水洗。无水硫酸钠干燥,减压真空下旋转蒸发得到黄色的混合物。硅胶柱层析法进行纯化,1.5倍样品拌样(硅胶100-200目),经层析柱分离(80 g的层析柱),用乙酸乙酯/石油醚(0~20%)的洗脱液梯度脱离,流速45 mL/min。得到7-溴-2-庚酮约17.5 g(84 mmol),收率91%,性状是黄色的油状物。经核磁氢谱和质谱进行确证,见图3。
3.2 注意事项
由于空气中的氧和水分与格氏试剂发生剧烈的氧化放热,易燃物。所以取用格氏试剂时要在氮气保护下进行,体系中尽量做到无水无氧。格式试剂不能长时间放置,活性会逐渐变低。
4 结论
(1) 化合物2对紫外、红外吸收都极弱,质谱极难表现出来,所以对其加入醇类化合物,使其化合物上的氯元素被醇取代从而生成酯类化合物,对紫外有吸收,便于分辨起始物是否反应完全。收率96%。
(2) 化合物3是弱碱性化合物,由化合物2在碱性条件下合成,化合物2加入反应体系时会放热,所以要在低温下滴加。
(3) 化合物4 在格氏试剂反应的条件下,由于加成产物结构相对稳定,就不会再与格氏试剂反应,这样就保证只上一个烃基。但在后处理加酸的条件下,这个中间产物变得不稳定,分解成酮和N-甲氧基甲胺盐。
(4)该路线具有工艺简单、操作方便及总收率高的优点,可适用于批量生产,其总收率78.6%。
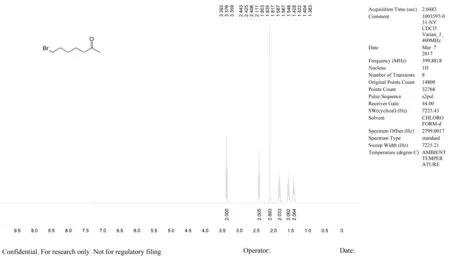
LC-MS (MS (ESI) m/z: 386.2 [M+M],tR= 1.091 min.1H NMR (400 MHz, CHLOROFORM-d) δ ppm 1.34 - 1.46 (m, 2 H) 1.57 (quin, J=7.55 Hz, 2 H) 1.84 (quin, J=7.11 Hz, 2 H) 2.09 - 2.14 (m, 3 H) 2.42 (t, J=7.39 Hz, 2 H) 3.38 (t, J=6.73 Hz, 2 H)
图3 7-溴-2-庚酮图谱
[1] 杜碧莹,王 自.己酮可可碱葡萄糖注射液中杂质的研究[J].中国药学杂志, 2014, 49(9):773-775.
[2] 万 嵘,王国伟,曹 玲,等. 7-溴-2-氧代庚酸的合成[J].中国医药工业杂志, 2005, 36(9): 531-532.
[3] 周红雨, 董为伟. 预防性治疗急性实验性自身免疫性脑脊髓炎的实验研究[J]. 华西医科大学学报, 2002, 33(3): 404-406, 436.
[4] 杨辉荣, 黎碧娜, 林自强, 等. 醛、酮α-溴代反应的研究过溴型聚合物试剂的合成及应用[J].化学试剂, 1989 (05): 267-269, 283.
[5] 张 玲,窦学杰, 赵桂森. 6-溴-2-己酮的合成[J].齐鲁药事, 2006, 25(3): 168-169.
[6] 梁文锦. 《化工产品应用手册》[J]. 广州化工, 1988 (01): 52.
[7] 张文龙, 陈希铭, 赵春山, 等. 对硝基苯胺接枝1,8-萘酰亚胺聚磷腈合成及表征[J].哈尔滨理工大学学报, 2015, 20(4): 30-34.
[8] Tomimatsu T,Murakami K.Novel steroid saponins,process for their extract ion and process for preparing 16-dehydropregnenolone therefrom:WO,8301065[P]. 1983-03-31.
(本文文献格式:杜远新,罗树常.7-溴-2-庚酮的合成[J].山东化工,2017,46(08):-.)
Improved synthesis of 7-bromoheptan-2-one
DuYuanxin1,LuoShuchang1*
(1. School of Chemical Engineering, Guizhou University of Engineering Science, Bijie 551700, China)
7-bromoheptan-2-one was synthesized starting from 6-bromohexanoic acid by a series of acyl-chlorination, Weinreb acylation and the displacement by Grignard reagent. The structures of the intermediates and final product were characterized by HNMR and ESI-MS. This modified method has advantages of simple process and high yield.
7-bromoheptan-2-one; 6-bromohexanoic acid; improved synthesis
2017-03-16
杜远新(1992—),本科生,主要从事药物中间体合成;通信作者: 罗树常(1984—),讲师,主要从事功能材料合成。
O622.2
B
1008-021X(2017)08-0055-03
猜你喜欢
现代仪器与医疗(2022年4期)2022-10-08 05:54:40
大学化学(2021年2期)2021-04-09 11:15:32
世界农药(2019年3期)2019-09-10 07:04:16
世界农药(2019年3期)2019-09-10 07:04:14
铜仁学院学报(2018年6期)2018-07-05 09:47:36
中成药(2017年4期)2017-05-17 06:09:50
百姓生活(2016年6期)2016-06-22 14:39:00
实验室研究与探索(2015年3期)2015-02-21 06:26:55
火炸药学报(2014年3期)2014-03-20 13:17:38
华东理工大学学报(自然科学版)(2014年1期)2014-02-27 13:48:32
