
Mitochondrial homeostasis in Parkinson’s disease - a triumvirate rule?
2017-09-04 07:27Chee-HoeNg,LitingHang,Kah-LeongLim
中国神经再生研究(英文版) 2017年8期
Mitochondrial homeostasis in Parkinson’s disease - a triumvirate rule?
Mitochondrial dysfunction in Parkinson’s disease: Mitochondria are the primary energy generator of the cell and they are important for cell survival and apoptosis. Defective mitochondrial homeostasis is frequently reported in human diseases especially those affecting the brain. Parkinson’s disease (PD), a prevalent neurodegenerative disorder where patients progressively loss control of their movements, is linked to mitochondrial dysfunction. A9-type dopaminergic (DA) neurons in the substantia nigra pars compacta, are signif i cantly diminished in PD brains. These neurons are known to be more susceptible to oxidative stress and mitochondrial toxins compared to other neuronal subtypes. Indeed, exposure to herbicides and pesticides such as paraquat and rotenone are linked to sporadic PD and represents a popular strategy to generate animal models of PD. Furthermore, studies carried out in induced pluripotent stem cells (iPSCs) conf i rmed the relevance of mitochondrial dysfunction and oxidative stress in PD. Interestingly, familial PD cases are linked to genes with apparent diverse functions that nonetheless converge towards the mitochondrion.ese PD-linked genes interact closely with each other through their participations in pathways necessary for the maintenance of mitochondrial homeostasis. For example, α-synuclein (α-syn) and leucine-rich repeat kinase 2 (LRRK2) could regulate mitochondrial fission/fusion. DJ-1, an oxidative stress sensor and protease, localizes to the mitochondria during oxidative stress.e ubiquitin ligase Parkin and mitochondrial serine kinase PTEN-induced putative kinase 1 (PINK1) collaborate to execute mitochondrial quality control through mitophagy, a process whereby damaged mitochondria are removed selectively.e mitochondrial serine protease high temperature requirement A2 [(HtrA2)/ Omi] protein acts downstream of PINK1 but in a pathway parallel to Parkin. Interestingly, aberrant mitochondrial fragmentation is often associated with autosomal dominant Parkinsonism caused by mutations in α-syn and LRRK2. On the other hand, autosomal recessive mutations of Parkin, PINK1, DJ-1 and ATPase type 13A2 (ATP13a2) tend to give rise to mitochondrial swelling phenotypes. In essence, we could appreciate the intimate relationship between mitochondrial dysfunction and PD pathogenesis. Given this, unravelling the mechanisms underlying mitochondrial abnormalities holds promise to enhance our understanding of the disease etiology and the development of ef f ective treatment strategies for PD.
AMPK- the chief orchestrator of mitochondrial homeostasis? AMP-activated protein kinase (AMPK), a serine/ threonine kinase, is a well-known intracellular master energy sensor whose activity is tightly linked to the level of adenosine monophosphate (AMP) and adenosine triphosphate (ATP) in the cell.rough this ability, AMPK actively monitors the catabolic and anabolic activity state of the cell. When ATP is abundant, AMPK activity is kept low. When energy is depleted such as during starvation, AMPK is activated by the elevated AMP level, followed by phosphorylation of the enzyme by its upstream kinases such as liver kinase B1 (LKB1), calcium/calmodulin kinase kinase II (CAMKKII) or transforming growth factor beta-activated kinase 1 (TAK1). Although AMPK has many important substrates, our discussion will focus on those linked to mitochondrial homeostasis. At least four known AMPK substrates, mitochondrial fi ssion factor (Mf f), dynamin-related protein 1 (Drp1), unc-51-like kinase 1 (Ulk1) and PPARγ coactivator-1 α (PGC-1α), are the main executioners of mitochondrial processes such as fi ssion, mitophagy and biogenesis. Activated AMPK could catalyze the phosphorylation of Mf f at serine155and serine172thereby recruiting Drp1 to activate fission (Toyana et al., 2016) Conversely, the enzyme could inhibit mitochondrial fi ssion by directly phosphorylating Drp1 at serine637. AMPK also regulates autophagy by phosphorylating Ulk1 at serine317and serine777or coordinates with protein deacetylase sirtuin 1 (Sirt1) to induce mitochondrial biogenesis by phosphorylating PGC-1α at serine177and serine538. Furthermore, these processes oen crosstalk with each other, for example inducing PGC-1α to suppress mitochondrial fission, and activating fissionviaMff/Drp1 is pivotal to mitophagy (Peng et al, 2016; Toyama et al., 2016). As summarized in Figure 1, AMPK is pivotal in coordinating metabolic fate of mitochondria in neuronsviaa triumvirate rule: modulating mitophagy, fission and biogenesis according to cellular energy requirements. Interestingly, PD gene mutations tend to result in dysregulation of these processes, suggesting that AMPK is a potential therapeutic target for PD. Indeed, pharmacological activators of AMPK such as 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR) and Metformin exhibit some success in improving Parkinsonian phenotypes in experimental and toxin-induced models of PD although conf l icting results pointing towards the detrimental effects of AMPK activation were also observed.
Mitochondrial fission is primarily controlled by Drp1, a small cytosolic GTPase. Drp1 translocates to the mitochondria and associates with one of its mitochondrial-bound receptors such as Fis1 or Mf f to catalyze membrane separation during fission. Pro-fusion GTPases such as Opa1 and Mitofusin 1/2 act in an opposite fashion to fi ssion machineries by mediating mitochondrial fusion. AMPK ameliorates mitochondrial fi ssion directlyviainhibiting Drp1 activity. Alternatively, AMPK can activate mitochondrial fi ssion during autophagy induction through phosphorylating Mff and recruitment of Drp1 (Toyama et al., 2016).us, AMPK plays a Janus-like role in regulating mitochondrial fission. Both fi ssion and fusion play equally important roles in neurons in terms of mitochondrial homeostasis and ATP synthesis. Mitochondrial fi ssion is critical for mitophagy and also for mitochondrial motility. As smaller mitochondria are more mobile, mitochondrial fi ssion ensures that the organelle are of the appropriate size to be traf fi cked along the axons. Indeed, loss of Drp1 in DA neurons leads to axonal degeneration as a result of the reduction in mitochondrial mass at the synapses due to impaired traf ficking of mitochondria (Berthet et al., 2014). Mitofusin 1 and 2 are targets of PINK1/Parkin-dependent ubiquitination during mitophagy, probably as a way to fuse damaged mitochondria to endoplasmic reticulum (ER) or lysosome for degradation (Gegg et al., 2010). Pharmacologically and genetically inhibiting Drp1 has been shown to prevent dopaminergic neuronal loss and restore dopamine release in PINK1-deficient mice. Familial LRRK2 point mutation G2019S increased Drp1 phosphorylation at thre-onine575resulting in hyperf i ssion and augmented autophagy (Su et al., 2013). Inhibiting Drp1 activity using mimetic peptide or dominant negative Drp1 mutant could reverse mitochondrial dysfunction and protect against degeneration of DA neurons derived from iPSCs from this patient. Overall, inhibiting fi ssion or ameliorating augmented autophagy seems to be rather promising approaches against certain forms of PD.
PGC-1α is important for the survival of DA neurons. Conditional knockout of PGC-1α leads to the reduction in DA neuron (Jiang et al., 2016). We have reported similar observation inDrosophila spargelmutant, an ortholog to mammalian PGC-1α (Ng et al., 2017). These spargel mutants exhibit marked reduction in PGC-1α mRNA transcripts, exhibit age-dependent loss of mobility and signif i cant depletion of dopamine due to degeneration of DA neurons. Interestingly, over-expression PGC-1α could also modulate the protein level as well as the activity of fi ssion/ fusion proteins Drp1 and Mfn2 in PC12 cells treated with rotenone, which suggests that a crosstalk between mitochondrial biogenesis and fi ssion/fusion machineries (Peng et al., 2016).
Defective mitochondrial biogenesis in PD: PGC-1α is a master regulator of mitochondrial biogenesis. Although the role of PGC-1α in skeletal muscle autophagy and mitochondrial biogenesis had been well studied, far less was done in term of understanding its roles in neurons. PGC-1α has been reported to be remarkably downregulated in PD patients, which implies a relationship between mitochondrial biogenesis and PD. Conditionally knocking out PGC-1α in adult mousebrain results in the loss of dopaminergic neurons (Jiang et al., 2016). A study done usingDrosophilaPGC-1α homologspargelyields similar conclusion (Ng et al., 2017).
A novel relationship between Parkin and PGC-1α was fi rst reported by Shin et al. in 2011. In this report, the investigators discover Parkin’s role in regulating PGC-1α transcription. Parkin ubiquitinates Parkin interacting substrate (PARIS/ZNF746), a repressor of PGC-1α, targeting it for proteasomal degradation. In the absence of Parkin, PARIS is stabilized and bound to the promoter of PGC-1α, switching of f PGC-1α expression that results in DA neuronal death (Figure 2).
Three separate studies also further established that transcriptional repression of PGC-1α as a mechanism underlying neurotoxin effects and neuroinflammation in PD. Siddiqui et al. in 2012 reported that mitochondrial toxin manipulates mitochondrial biogenesis in 2 ways, increasing α-syn-dependent repression as well as decreasing myocyte enhancer factor 2C (MEF2C)-mediated transcription of PGC-1α, with oxidative stress exerting a synergistic ef f ect. Oxidative stress enhances α-syn-dependent repression and S-nitrosylation of MEF2C leading to decreased biogenesis. Furthermore, the investigators observed that mutant α-syn A53T is twice as ef fi cient in repressing the promoter of PGC-1α compared to wild-type α-syn.is conclusion was also supported by another study whereby the investigators observed DA neurons in PGC1α-KO mice show abnormal mitochondria and fragmented endoplasmic reticulum and further expression of human α-syn exacerbated degeneration of these vulnerable neurons (Ciron et al., 2015).
To understand the impact of neuroinf l ammation, a prominent feature of PD, the pro-inflammatory fatty acid, palmitate was administered toex vivocultured primary mouse cortical neurons, microglia and astrocytes. Palmitate-treated cells showed significant cytosine methylation in PGC-1α promoter resulting in reduced mitochondrial content (Su et al., 2015). Similarly intracerebroventricular injection of palmitate into mice overexpressing human α-syn resulted in enhanced methylation of PGC-1α promoter, decreased PGC-1α expression and reduced mitochondrial content in substantia nigra.
Last but not least, the level of PGC-1α was signif i cantly decreased in mnd2 (motor neuron degeneration 2) mouse, a mutant mouse strain harboring homozygous recessive mutation in Omi (Xu et al., 2014). Mnd2 mice exhibit neurodegeneration, mitochondrial abnormalities, stunted growth and rarely survived beyond 40 days postnatally. Levels of NRF-1 and TFAM mRNA which are targets of PGC-1α, were found to be markedly reduced in mnd2 mouse brain compared with wide-type suggesting that Omi may be associated with the reduction of PGC-1α. Omi was found to cleave glycogen synthase kinase 3β (GSK3β), and the latter can phosphorylate PGC-1α and promote its ubiquitin-mediated degradation.
Conclusions: We have discussed here the participation of several mitochondrial pathways such as fission, mitophagy and biogenesis in the pathogenesis of PD. Given that seemingly diverse genes associated with PD all converged at the mitochondria, one could envisage the possibility to counteract neurodegeneration, especially in the case of PD,viamitochondrial-related therapeutics. In so far, most results suggest that the mitochondrial pathology in PD may be due to the imbalance of the mitochondrial dynamics or biogenesis, the latter clearly illustrates the importance of PGC-1α in DA neuronal survival. Notably, two animal studies, one in rodent and the other in Drosophila, robustly support the importance of PGC-1α in DA neuronal survival. Transcriptional repression of PGC-1α promoter is another importantpathological mechanism that exemplif i es the role of PGC-1α in PD. Further investigations are needed to see if a common pathway such as those regulated by AMPK or its substrates could be tapped for treating PD.
Chee-Hoe Ng*, Liting Hang, Kah-Leong Lim
National Neuroscience Institute, Singapore (Ng CH, Hang L, Lim KL)
Neuroscience & Behavioral Disorders Program, Duke-NUS Medical School, Singapore (Lim KL)
Department of Physiology, National University of Singapore, Singapore (Lim KL)
NUS Graduate School for Integrative Sciences and Engineering, Singapore (Hang L)
*Correspondence to: Chee-Hoe Ng, Ph.D., CheeHoe267@yahoo.com.sg.
orcid: 0000-0001-8545-0497 (Chee-Hoe Ng) 0000-0002-5440-2588 (Kah-Leong Lim)
Accepted:2017-07-24
How to cite this article:Ng CH, Hang L, Lim KL (2017) Mitochondrial homeostasis in Parkinson’s disease - a triumvirate rule? Neural Regen Res 12(8):1270-1272.
Plagiarism check: Checked twice by ienticate.
Peer review: Externally peer reviewed.
Open access statement:is is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build uponthe work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.

Figure 1 PD genes interact with mitochondrial fi ssion, biogenesis and mitophagy.
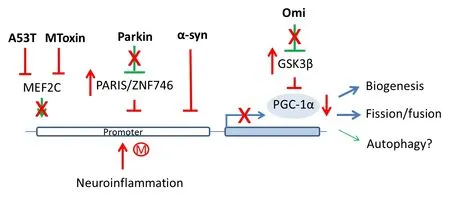
Figure 2 Transcriptional repression of PGC-1α by PD genes, mitochondrial insults and neuroinf l ammation.
Open peer reviewers: Rui Silva, Universidade de Lisboa Faculdade de Farmacia, Portugal; Jin Zhang, Dalhousie University, Canada.
Berthet A, Margolis EB, Zhang J, Hsieh I, Zhang J, Hnasko TS, Ahmad J, Edwards RH, Sesaki H, Huang EJ, Nakamura K (2014) Loss of mitochondrial fission depletes axonal mitochondria in midbrain dopamine neurons. J Neurosci 34:14304-14317.
Ciron C, Zheng L, Bobela W, Knott GW, Kelly DP, Schneider BL (2015) PGC-1α activity in nigral dopamine neurons determines vulnerability to α-synuclein. Acta Neuropathol Commun 3:16.
Gegg ME, Cooper JM, Chau KY, Rojo M, Schapira AH, Taanman JW (2010) Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum Mol Genet 19:4861-4870
Jiang H, Kang SU, Zhang S, Karuppagounder S, Xu J, Lee YK, Kang BG, Lee Y, Zhang J, Pletnikova O, Troncoso JC, Pirooznia S, Andrabi SA, Dawson VL, Dawson TM (2016) Adult conditional knockout of PGC-1α leads to loss of dopamine neurons. eNeuro 3:ENEURO.0183-16.2016.
Ng CH, Basil AH, Hang L, Tan R, Goh KL, O’Neill S, Zhang X, Yu F, Lim KL (2017) Genetic or pharmacological activation of the Drosophila PGC-1α orthologspargel rescues the disease phenotypes of genetic models of Parkinson’s disease. Neurobiol Aging 55:33-37.
Peng K, Yang L, Wang J, Ye F, Dan G, Zhao Y, Cai Y, Cui Z, Ao L, Liu J, Zou Z, Sai Y, Cao J (2016)e interaction of mitochondrial biogenesis and fi ssion/ fusion mediated by PGC-1α regulates rotenone-induced dopaminergic neurotoxicity. Mol Neurobiol 54:3783-3797.
Ryan SD, Dolatabadi N, Chan SF, Zhang X, Akhtar MW, Parker J, Soldner F, Sunico CR, Nagar S, Talantova M, Lee B, Lopez K, Nutter A, Shan B, Molokanova E, Zhang Y, Han X, Nakamura T, Masliah E, Yates JR 3rd, et al. (2013) Isogenic human iPSC Parkinson’s model shows nitrosative stress-induced dysfunction in MEF2-PGC1α transcription. Cell 155:1351-1364.
Shin JH, Ko HS, Kang H, Lee Y, Lee YI, Pletinkova O, Troconso JC, Dawson VL, Dawson TM (2011) PARIS (ZNF746) repression of PGC-1α contributes to neurodegeneration in Parkinson’s disease. Cell 144:689-702.
Siddiqui A, Chinta SJ, Mallajosyula JK, Rajagopolan S, Hanson I, Rane A, Melov S, Andersen JK (2012) Selective binding of nuclear alpha-synuclein to the PGC1alpha promoter under conditions of oxidative stress may contribute to losses in mitochondrial function: implications for Parkinson’s disease. Free Radic Biol Med 53:993-1003.
Su X, Chu Y, Kordower JH, Li B, Cao H, Huang L, Nishida M, Song L, Wang D, Federof f HJ (2015) PGC-1α promoter methylation in Parkinson’s disease. PLoS One 10:e0134087.
Su Y, Qi X (2013) Inhibition of excessive mitochondrial fi ssion reduced aberrant autophagy and neuronal damage casued by LRRK2 G2019S mutation. Human Mol Genet 22:4545-4561.
Toyama EQ, Herzig S, Courchet J, Lewis TL Jr, Losón OC, Hellberg K, Young NP, Chen H, Polleux F, Chan DC, Shaw RJ (2016) AMP-activated protein kinase mediates mitochondrial fi ssion in response to energy stress. Science 351:275-281.
Xu R, Hu Q, Ma Q, Liu C, Wang G (2014)e protease Omi regulates mitochondrial biogenesis through the GSK3β/PGC-1α pathway. Cell Death Dis 5:e1373.
10.4103/1673-5374.213546
- 中国神经再生研究(英文版)的其它文章
- Transcriptional inhibition in Schwann cell development and nerve regeneration
- A progressive compression model of thoracic spinal cord injury in mice: function assessment and pathological changes in spinal cord
- Effects of estrogen receptor modulators on cytoskeletal proteins in the central nervous system
- Optogenetics and its application in neural degeneration and regeneration
- Live-cell imaging: new avenues to investigate retinal regeneration
- Neurotrophic factors and corneal nerve regeneration