
基于SSR标记的楸树遗传多样性及核心种质构建1)
2017-09-03 09:22方乐成夏慧敏
东北林业大学学报 2017年8期
方乐成 夏慧敏
(南京林业大学,南京,210037)
麻文俊
(中国林业科学研究院林业研究所)
张新叶
(湖北省林业科学研究院)
基于SSR标记的楸树遗传多样性及核心种质构建1)
方乐成 夏慧敏
(南京林业大学,南京,210037)
麻文俊
(中国林业科学研究院林业研究所)
张新叶
(湖北省林业科学研究院)
利用SSR标记对192个楸树种质资源进行遗传多样性和亲缘关系研究。试验筛选出13对引物对192份供试材料进行扩增,共获得89个等位基因位点,有效等位基因平均为3.795 9,Shannon’s多样性指数平均值为0.506 6;Nei’s遗传多样性平均值为0.667 7。用MEGA6.0软件对192份楸树材料进行遗传距离分析,通过聚类分析构建出供试材料楸树种质资源间的聚类图。利用SSR分子标记,采用多次聚类结合位点优先的取样策略,比较了样本数不同的4个核心样本群的等位基因数、有效等位基因数、Shannon’s指数和Nei’s遗传多样等参数,初步构建了192份楸树种质材料的46份核心种质。核心种质保留了初始种质23.96%的样品。
楸树;微卫星;分子标记;遗传多样性;核心种质
楸树(CatalpabungeiC.A.Mey.)属紫葳科梓树属,落叶乔木,原产中国,已有三千多年的栽培历史,是中国特有的珍贵用材树种以及著名的园林观赏树种[1]。由于其材质优良、适应性强、用途广泛、生长快,自古便有"木王"的美称,是用于建立复合农林业建设的理想树种[2]。由于楸树自花不孕,种子发芽率低,扦插生根困难,再加上人们对它的长期采伐利用,导致楸树资源破坏严重[3]。因此,全面了解楸树资源状况,构建核心种质,对我国楸树资源的合理开发利用有重要的理论价值和实践意义。
核心种质是种质资源的核心子集,能够通过最少数的遗传资源,最大限度地保存整个资源群体的遗传多样性[4]。Frankel[5]首先提出核心种质的概念并将其作为研究种质资源的关键。由于基于形态学的传统研究思路和方法存在很多先天不足,种质资源基础研究一直效率低下,随着分子标记和测序技术的快速发展,基因组学理论和研究方法不断深入到种质资源研究领域中,为种质资源的研究方法开拓了新的思路[6]。运用分子标记可以阐述物种起源演化,评估资源基因结构多样性,而新一代测序技术[7]的应用能够进行基因挖掘并且在全基因组水平上比较不同种质资源的基因组差异。鉴于此,本研究选用SSR标记对我国不同地区的楸树资源进行研究,分析其遗传多样性,目的在于对楸树进行核心种质的初步构建并为楸树种质资源的收集利用和新品种选育提供理论依据。
1 材料与方法
1.1供试材料
供试楸树资源共192份,其中包括洛阳楸树基因库资源85份,南阳楸树基因资源64份,偃师楸树基因资源15份,鄂西北楸树基因资源28份。选取各楸树资源幼嫩叶片5~6片,用75%酒精擦拭后,放入冰盒带回实验室,于冰箱-80 ℃保存,用于DNA样品提取。
1.2 分子标记
DNA提取:采用改良的CTAB-SDS结合法提取楸树叶片DNA,用1%的琼脂糖凝胶以及ND-2000紫外分光光度仪检测DNA浓度,合格的DNA样品保存于-20 ℃用于后续实验。
SSR引物筛选:根据中国林科院林业所提供的3 646个SSR位点信息,应用引物设计软件Primer premier5.0共设计出85对引物,由上海捷瑞公司合成。为了得到清晰的SSR指纹图谱,随机选取6个楸树DNA样品作为模板DNA,从合成的85对引物中筛选出13对效果较好的引物,具体信息见表1。

表1 楸树SSR引物序列及特征
SSR分析:利用筛选出来的13对引物对192个DNA样品进行含荧光碱基的PCR反应,产物交由美吉生物公司用ABI3730毛细管电泳检测多态性,原始数据运用genemapper v3.7软件分析。
1.3 数据分析
使用POPGENE V1.31软件计算Nei’s遗传距离[8]、有效等位基因数、期望杂合度、观测杂合度、扩增位点的多态性信息量以及香农信息指数等指标评价13对引物在192个楸树资源中的遗传多样性。运用非加权算数平均法(UPGMA),对192份材料进行聚类分析,并在MEGA6.0软件中显示出聚类图[9]。
1.4 核心种质构建
本试验采取多次聚类,以随机取样策略为对照策略[10],应用位点优先的取样方法[11]。通过UPGMA方法进行遗传距离分析,用MEGA6.0软件构建聚类图,根据树形图,从分类水平最低的两个相近的遗传材料中选择具有最多稀有等位基因数的材料,如果两个相近的遗传材料具有相等数目的稀有等位基因,则优先选择稀有等位基因频率值较小的,如果这两个值仍然相同则随机选择,组内只有一个材料的直接选入,对所取出的材料再次聚类,直到取得的遗传材料数量占总数的20%~30%,构成核心种质的样本群[12]。
1.5 核心种质评价
对核心种质和初始种质的有效等位基因数、Shannon’s多样性信息指数、遗传多样性等指标进行t检验,以此评价核心种质的代表性。
2 结果与分析
2.1 SSR分子标记遗传多样性
采用SSR标记对192份楸树资源的多样性进行分析。13对SSR引物共检测到89个等位基因,其中88个为多态性等位基因,多态率达99%,每对引物获得的等位基因数从3(28和47号引物)到16(62号引物)个不等,平均6.85个,扩增所得的产物大小在150~500 bp。
扩增结果发现,有的楸树资源产生了特有的条带,可作为重要的分子性状用于品种鉴定。192份楸树资源经13对引物扩增产生的89条多态性谱带中,特有性谱带有4条,占4.49%。其中,55号引物在基因资源“9.2.3(鄂西北)”上有一条特有带,79号引物也在该资源上有一条特有带;而70号引物在基因资源“西峡8号”和“7.1(鄂西北)”上各有一条特有谱带(表2)。

表2 192份楸树种质的特有SSR标记
运用13对引物对各楸树样品的STR分型数据,结合POPGENE软件的数据类型,用英文字母表示不同的带型,形成可鉴定各品种的特征指纹代码。
2.2 楸树种质的聚类抽样
运用POPGENE软件分析192份楸树种质资源的遗传多样性。192份楸树资源的Shannon’s指数平均为0.506 6,有效等位基因平均为3.795 9,Nei’s遗传多样性的值平均为0.667 7。从群体间遗传多样性分析来看:洛阳的Shannon’s指数和Nei’s遗传多样性最高,分别为0.498 3和0.649 7;其次是鄂西北,分别是0.475 8和0.614 4;南阳是0.492 7和0.587 5;偃师最低,Shannon’s指数和Nei’s遗传多样性量分别是0.459 3和0.593 6。
对192份楸树材料进行遗传距离分析,用MEGA 6.0构建出这192份供试材料的聚类图(图1)。该聚类图能够很好地体现了各材料之间的亲缘关系。根据聚类图,大致可将192份楸树资源可分为4大类。
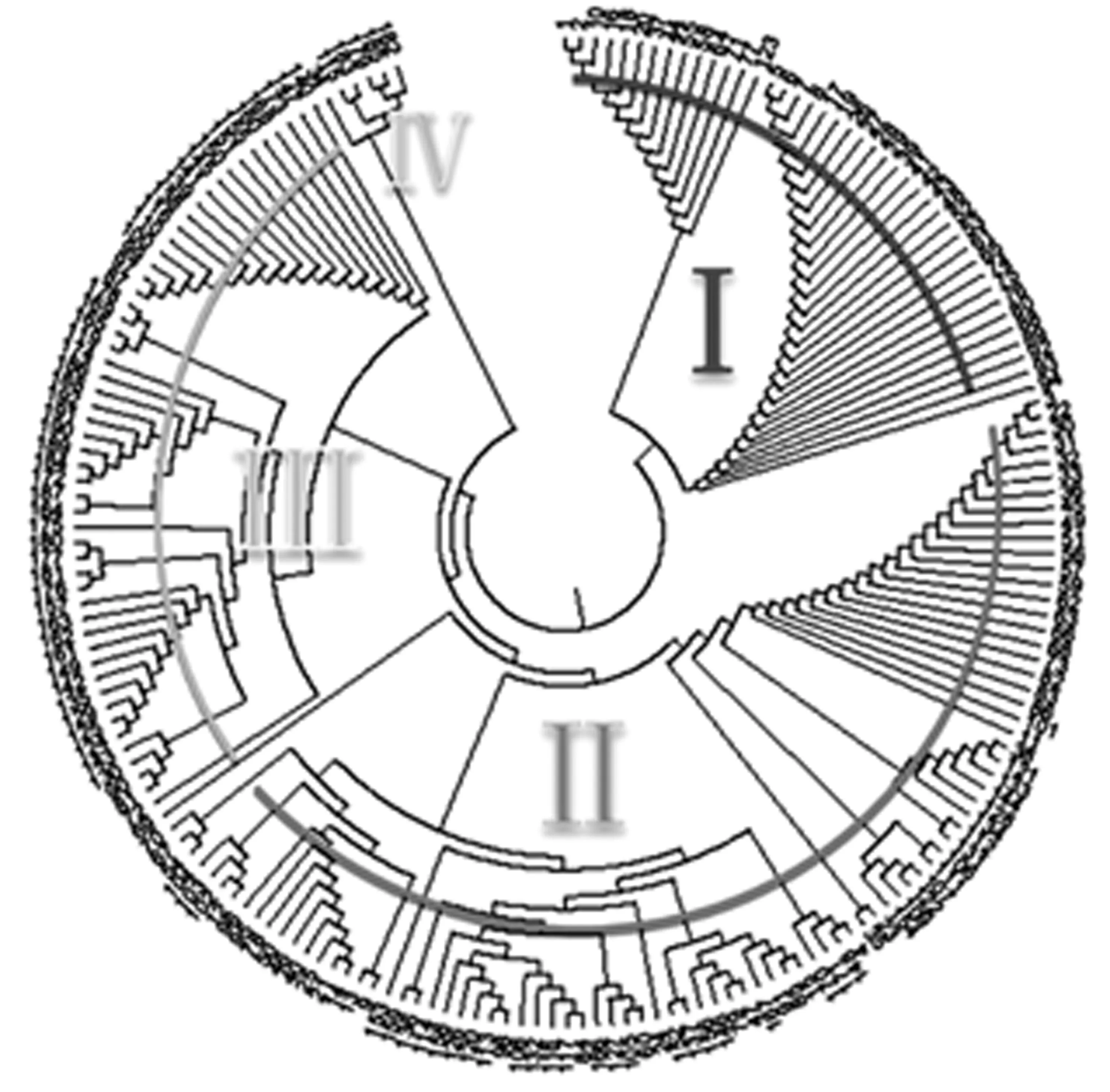
图1 192份楸树种质资源的聚类分析
第一类(Ⅰ)包括41份楸树种质资源,其中洛阳39份(1012等),占第一类资源的95.1%,偃师1份(7-5),南阳1份(新野4号),各约占第一类的2.44%;第二类(Ⅱ)包括91份楸树种质资源,其中洛阳42份(3103等),占46.15%,南阳35份(镇平3号等),占第二类的38.46%,偃师12份(3-1等),约占13.19%,鄂西北2份(9.1,8.1),占第二份总数的2.2%;第三类(Ⅲ)包括52份楸树资源,其中洛阳3份(6019,1049,3422),占第三类总数的5.77%,南阳27份(镇平1号等),占第三类总数的51.92%,鄂西北22份(7.1等),约占第三类总数42.3%;第四类(Ⅳ)6份楸树种质资源,其中洛阳1份(9008),南阳1份(淅川2号),各占第四类的16.67%,鄂西北4份(9.2.3,9.3.1,9.5.2,13.1),占第四类总数的66.67%。

表3 样品群间的遗传多样性比较
全部192个楸树样品,经过4次聚类抽样,最后得到一个由46个样品组成的核心样本群,并用等位点数、有效等位基因数、Nei’s遗传多样性、Shannon’s指数等指标评价4个样品群的遗传多样性。
经过统计,样品群4的等位点数、有效等位基因数、Shannon’s指数、Nei’s遗传多样性值等指标均是最高,可以作为最后的核心种质。
核心种质保留了初始种质23.96%的样品,其中有效等位基因数为84,保留率为94.38%;有效等位点数为3.934 3,保留率为103.6%;Shannon’s指数为0.510 0,保留率为101.8%;Nei’s遗传多样性值为0.665 5保留率为99.7%。
46个核心种质:4018、9024、1008、1039、镇平3号、西峡3号、淅川9号、9.2.1、3212、2-1、8-5、9008、7-8、5016、JS1、7-5、6025、130、1117、4023、1-52、3422、4021、8-12、内乡1号、镇平5号、淅川3号、内乡13号、淅川6号、邓州3号、西峡8号、邓州17号、唐河2号、邓州18号、卧龙2号、卧龙1号、南召13号、9.1.3、7.4、7.1、5.6、9.5.3、9.3.1、9.2.3、12.5、12.1。其中洛阳种质资源15份,占核心种质总数的32.6%;南阳种质资源16份,占核心种质总数的34.8%;偃师种质资源5份,占总数的10.9%;鄂西北种质资源10份占核心种质总数的21.7%。
2.3 楸树核心种质有效性分析检验
对2个群体的有效等位基因、Nei’s遗传多样性、Shannon’s指数分别作t检验。从表4中t测验的结果可以看出核心种质的有效等位基因数、Nei’s遗传多样性、Shannon’s指数在概率0.05水平上与初始种质差异不显著,可见核心种质能很好的代表原始种质。
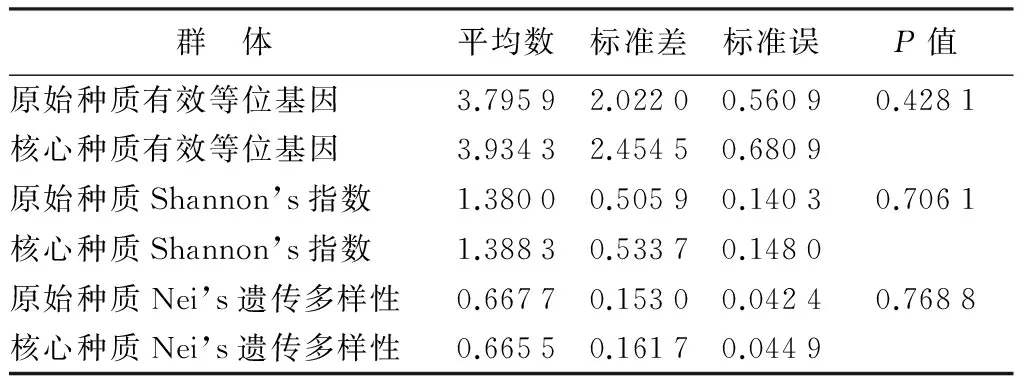
表4 初始种质和核心种质的t测试结果
保留种质是指去除核心种质后初始种质中剩余的部分,它可以作为核心种质的后备资源,当无法在核心种质中匹配到所需性状时,即可从其中寻找。将保留种质与核心种质在样品数,等位基因数、有效等位基因数、Nei’s遗传多样性、Shannon’s指数几个方面比较分析。
从表5可知核心种质的有效等位基因数、Shannon’s指数均高于保留种质,应该优先使用。
分别对保留种质的有效等位基因、Nei’s遗传多样性、Shannon’s指数进行t检验,结果如表6所示。从表6中可以看出,在概率0.05水平上保留种质与核心种质差异不显著,即核心种质较好的保留了全部种质的遗传多样性,丢失不显著。

表5 核心种质与保留种质遗传多样性比较
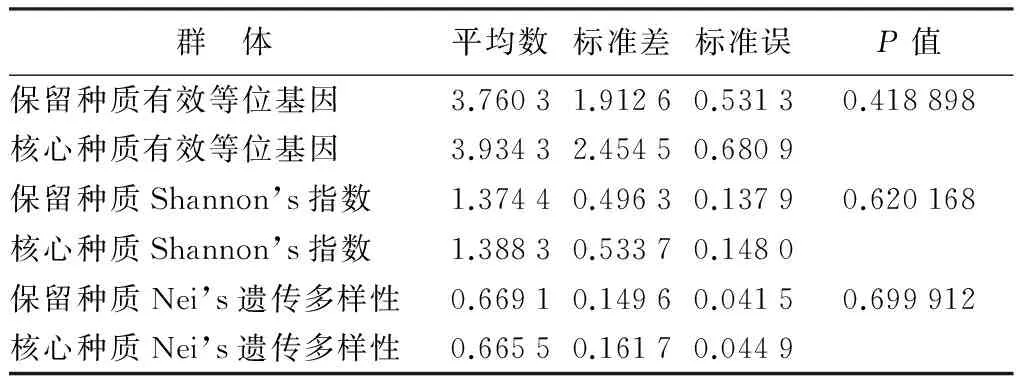
表6 保留种质和核心种质的t测试结果
3 结论与讨论
核心种质构建就是用科学的方法从整个种质资源中选出一部分样本,以最小的遗传资源数量来代表整个遗传资源的多样性,国内外不同植物核心种质库的构建,通常选择整个样本群的5%~30%,但总量不超过3 000份[14]。原始群体的大小和遗传结构是影响核心种质取样比例的关键因素,总资源多且遗传多样性小的物种,取样比例可以适当缩小,反之则增加取样比例[15]。本研究所使用的原始群体数量较少,同时为了尽量减少遗传多样性的丢失,将核心种质的总体取样比例设置在25%。
随着核心种质在遗传育种中的作用受到广泛的认同,大多数农作物和园林植物都展开了核心种质的构建工作。传统的核心种质构建方法,是比较不同种质资源在植物学、形态学、农艺、品质性状上的差异来剔除亲缘关系相近的样品并且抽取核心种质[15]。但是这些表型性状易受外界因素影响,尤其是数量性状受到气候、地理位置等环境因素影响而表现不稳定。分子标记数据不受环境等因素的影响,可以作为植物形态性状和植物生理特性的有效补充。AFLP、RAPD、SSR等分子标记技术广泛应用于育种工作,使育种工作者能从DNA水平上区分植物样品并构建核心种质[16]。
然而,由于分子生物学实验费用较高,对于种质资源原始群体庞大的物种,很难对每一个样品都用分子标记检测其多样性信息,董玉琛[17]认为大样本群的核心种质构建可以分两步进行,首先根据表型数据构建初选样品群,再对初选的样品群进行分子标记分析,最终建立核心种质。本实验由于总体样本量较少,所以直接采用分子标记分析的方法,但是扩大了取样比例。
目前,在分子水平上对林木进行核心种质构建研究较少,尤其是在楸树的种质资源构建方面,目前只有石欣等运用ISSR标记对10个类型的156个单株楸树进行了遗传分析[18],以及郝明灼等利用ISSR与SRAP结合的方法对138份楸树材料进行了分析[19]。本试验首次利用13对楸树SSR引物,对192份楸树种质资源进行扩增,13对引物共检测到89个等位基因,其中88个为多态性等位基因,多态百分率达99%。根据统计结果,洛阳群体的Shannon’s指数与遗传多样性最高,可能与样本数量以及洛阳的地理位置有关,由于洛阳样本数量最多,且洛阳地势西高东低,境内山川丘陵交错,地形复杂多样,全年气候四季分明,在适宜楸树生长的基础上一定程度的丰富了洛阳楸树资源的遗传多样性。
核心种质在构建中需要的不是最大化等位基因多样性,而是种质间的遗传距离,因为遗传距离越大,说明种质间差异越大,对育种越有利。本实验使用的多次聚类法正是基于Nei’s遗传距离,用UPGMA方法进行遗传距离分析,从分类水平最低的两个遗传材料中选出合适的样品,将种质间的遗传距离作为优先参考。
[1] 潘庆凯,郭明,康平生.关于发展楸树的若干意见[J].河南林业科技,1986(1):21-23.
[2] 郭从俭,钱士金,王团荣,等.楸树育苗技术研究[J].河南农业大学学报,1988,29(3):5743-5743.
[3] 彭方仁,郝明灼,梁有旺,等.我国楸树种质资源现状及开发利用策略[J].林业工程学报,2011,25(6):1-5.
[4] 王建成.构建植物遗传资源核心种质新方法的研究[D].杭州:浙江大学,2006.
[5] ARBER W, LLIMENSEE K, PEACOCK W, et al. Genetic Manipulation: Impact on Man and Society[M]. New York: Cambridge University Press,1984.
[6] 黎裕,李英慧,杨庆文.基于基因组学的作物种质资源研究:现状与展望[J].中国农业科学,2015,48(17):3333-3353.
[7] 张得芳,马秋月,尹佟明,等.第三代测序技术及其应用[J].中国生物工程杂志,2013,33(5):125-131.
[8] YEH F C, YANG R C, BOYLE T B, et al. Popgene the user-friendly shareware for population genetic analysis[M]. Edmonton: University of Alberta Canada,1997.
[9] TAMURA K, STECHER G, PETERSON D, et al. MEGA6: molecular evolutionary genetics analysis version 6.0[J]. Molecular Biology and Evolution,2013,30(12):2725-2729.
[10] HU J, ZHU J, XU H M. Methods of constructing core collections by stepwise clustering with three sampling strategies based on the genotypic values of crops[J]. Theoretical and Applied Genetics,2000,101(1):264-268.
[11] 张春雨,陈学森,张艳敏,等.采用分子标记构建新疆野苹果核心种质的方法[J].中国农业科学,2009,42(2):597-604.
[12] 胡晋,徐海明,朱军.基因型值多次聚类法构建作物种质资源核心库[J].生物数学学报,2000,15(1):103-109.
[13] HINTUM J, BROWN A D, SPILLANE C, et al. Core collection of plant cenetic resources[M]. London: Soyce Publishing,1995.
[14] BROWN A H D. Core collections: a practical approach to genetic resources management[J]. Genome,2011,31(2):818-824.
[15] KANG C W, KIM S Y, LEE S W, et al. Selection of a core collection of Korean sesame germplasm by a stpwise clustering method[J]. Breeding Science,2006,56(1):85-91.
[16] 邬荣领,尹佟明,黄敏仁,等.分子标记辅助选择在林木育种中应用[J].林业科学,2000,36(1):103-111.
[17] 董玉琛,曹永生,张学勇,等.中国普通小麦初选核心种质的产生[J].植物遗传资源学报,2003,4(1):1-8.
[18] 石欣,李亚,杨如同,等.中国楸树(CatalpabungeiC.A.Mey)种质资源遗传多样性的ISSR分析[J].江苏农业学报,2011,27(3):634-639.
[19] 郝明灼.中国楸树种质资源分布及遗传多样性分析[D].南京:南京林业大学,2013.
Genetic Diversity Analysis and Primary Core Collection ofCatalpabungeiGermplasm with SSR Markers//
Fang Lecheng, Xia Huimin
(Nanjing Forestry University, Nanjing 210037, P. R. China);
Ma Wenjun
(Research Institute of Forestry, Chinese Academy of Forestry);
Zhang Xinye
(Hubei Academy of Forestry)
//Journal of Northeast Forestry University,2017,45(8):1-5.
Constructing core collection is a useful way to improve efficiency of conserve and manage species germplasm. We used SSR markers for the genetic diversity analysis and genetic relationship research with 192Catalpabungeigermplasm, selected 13 pairs of SSR primers, amplified all of the 192 samples by these 13 pairs of primers, and found 89 alleles. The average effective number of allele (Ne) was 3.795 9, average of Shannon’s diversity index (I) was 0.506 6, and Nei’s genetic diversity (H) average number was 0.667 7. The analysis of genetic distance of 192C.bungeisamples with MEGA6.0, and the construction of the dendrogram ofCatalpasamples by cluster analysis was conducted. With SSR markers and the sampling strategy of multiple cluster binding loci preferential, we primarily constructed 4 core sample groups with different numbers. In these 4 groups, 46 core collection of the total 192 collectedCatalpatrees were compared by the parameters including the number of allele, effective number of allele, Shannon’s index and Nei’s genetic diversity. The core collections retained 23% of the original collections.
Catalpabungei; SSR; Molecular marker; Genetic diversity; Core collection
方乐成,男,1988年7月生,南京林业大学林学院,博士研究生。E-mail:651282772@qq.com。
张新叶,湖北省林业科学研究院,研究员。E-mail:1641135733@qq.com。
2017年1月25日。
S792.21
1)林业公益性行业科研专项(201404101)。
责任编辑:潘 华。
猜你喜欢
今日农业(2022年14期)2022-09-15
今日农业(2022年13期)2022-09-15
——致秋天的花楸树
北方人(2021年19期)2021-10-29
智慧健康(2021年17期)2021-07-30
中国产前诊断杂志(电子版)(2020年1期)2020-05-21
遵义医科大学学报(2020年6期)2020-02-05
山东林业科技(2019年2期)2019-06-03
中国麻业科学(2018年6期)2018-04-09
现代检验医学杂志(2016年5期)2016-08-20
广西林业科学(2016年3期)2016-03-16
