
先天性肌强直1例报告
2017-08-17 09:31:01顾平孙正芹王文婷韩瑞王大力
中国神经精神疾病杂志 2017年6期
顾平孙正芹王文婷韩瑞王大力
·病例报告·
先天性肌强直1例报告
顾平*孙正芹△王文婷*韩瑞*王大力△
先天性肌强直 CLCN1基因 基因变异
先天性肌强直 (myotonia congenital,MC)是与CLCN1基因相关的遗传性氯离子通道疾病,以肌肉收缩后舒张困难为主要表现,分为常染色体显性遗传的Thomsen型及常染色体隐性遗传的Becker型。目前,国外对MC相关CLCN1基因突变报道较多,而国内CLCN1基因确诊的MC仅有几个家系报告[1],且临床医师对此病缺乏足够的认识,容易漏诊。本文报告1例MC,对其临床特征及CLCN1基因进行探讨,以期提高临床工作者对此病的诊治能力。
1 临床资料
1.1 一般资料先证者,男,42岁,主因全身肌肉僵硬、动作笨拙30余年来我院就诊。患者诉自幼出现全身肌肉僵硬、动作笨拙,表现为久坐后起立困难,跑步启动费力,双手握拳后伸展困难,吃饭起始时张口费力,反复活动后上述症状减轻。劳累后可伴轻微肌无力,生气、温暖时肌强直症状加重,饮酒后、寒冷刺激症状减轻,紧张时无显著影响。无睁眼费力,无胸闷、活动后气促,无性功能减退,无二便异常。目前患者女儿7岁,儿子19岁,尚未出现类似症状。家族中患者姐姐有类似病史,余患者家属均未发现肌强直表现(图1)。
1.2 查体心率62次/min,心肺腹查体未见明显异常。神清语利,颅神经查体未见异常。双侧腓肠肌假性肥大,叩击可见肌球,四肢肌力5级,用力握拳后伸展缓慢,坐位后站起缓慢,肌张力正常,未发现不自主运动,病理征未引出,感觉、共济正常。
1.3 辅助检查肌电图提示肌强直电位。心电图:窦性心律,Ⅰ度房室传导阻滞。肌酶学:LD/HBDH:1.72(正常值:1.2~1.6),谷草转氨酶、乳酸脱氢酶、羟丁酸脱氢酶、肌酸激酶、肌酸激酶同工酶未见异常。凝血系列、乙肝、梅毒、艾滋抗体未见异常。头CT未见异常。神经传导速度未见异常。
1.4 基因分析先证者CLCN1基因发现c.1129C>T(编码区第1129号核苷酸由C变为T)的杂合核苷酸变异,该变异导致编译第377号氨基酸Arg的密码子变为终止密码子(p.Arg377Ter),从而使肽链合成提前终止,为无异变异;c.1887delC(编码区第1887号核苷酸C缺失)的杂核苷酸变异,该变异导致从第630号氨基酸开始的氨基酸合成发生改变(p.630fs),为移码变异。先证者儿子、女儿第1129号核苷酸未发现变异,第1887号核苷酸为CLCN1基因杂合子(图2)。
2 讨论
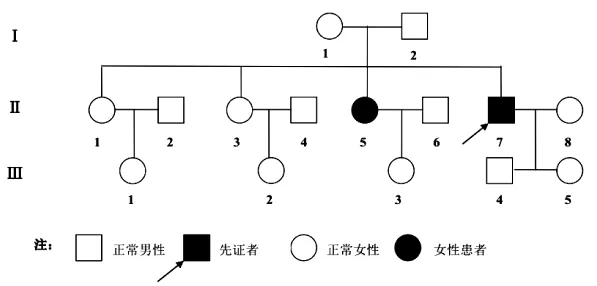
图1 先天性肌强直家系谱图
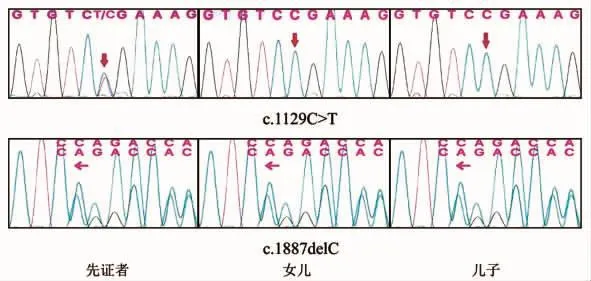
图2 先证者及其子女基因检测图。先证者存在c.1129C→T及c.1887C缺失突变;先证者子女存在c.1887C缺点突变,未发现c.1129C→T变异
MC是由于CLCN1基因突变导致的骨骼肌氯离子通道疾病,主要表现为常染色体显性遗传的Thomsen型及常染色体隐性遗传的Becker型。肌肉僵硬、“热身现象”(即随意运动后出现肌强直,活动后肌强直症状减轻)[2]与不同程度的肌肉肥大为二者的共同临床特征[3]。一般情况下,Thomsen型的临床症状发生在婴儿早期,上肢通常受累,与Becker型相比肌僵硬症状不明显,预后相对较好,肌无力与肌萎缩症状不明显[4]。而Becker型通常发生在一个人一生中第二个十年,通常下肢先受累,肌强直症状相对较严重,可发生永久性肌无力,但平滑肌和心肌不受影响。本例报道先证者十余岁起病,四肢同时受累,存在肌强直与“热身现象”,肌无力症状轻微,不影响日常工作和生活,未见明显肌萎缩,临床表型难以区分。此外,本研究先证者温暖环境可使肌强直症状加重,与FERRIBY等[5]的研究报道一致。
目前,国外c.1129C>T的致病性已经文献报道,与先天性肌强直相关[3],变异 c.1887delC的致病性尚未见文献报道。本例先证者存在 c.1129C>T变异,其子女未发现c.1129C>T,可初步推断本研究的遗传类型为常染色体隐性遗传,即Becker型。本例先证者及子女的c.1887delC变异类型为杂合,但其子女不存在先天性肌强直的临床特征,亦可推断其隐形遗传特性。肌强直药物试验发现,阻断50%的生理性氯电流不足以产生强直性活动,可解释隐性突变杂合携带者尽管氯电流下降50%,但临床表现不出现肌强直[6],支持本研究家系的临床特点。结合MC的遗传特点、临床特征、肌电图、肌肉活检及基因检测可诊断本病,给予本例患者心律平100 mg每日3次治疗有效。
迄今报道的CLCN1基因突变多达160多个[7],包括点突变、移码突变、剪接突变等。本研究发现先天性肌强直c.1129C>T、c.1887delC变异,对此家系MC产前诊断具有重大意义。此外,有待更多的遗传学研究丰富CLCN1致病基因谱系,为先天性肌强直的诊断与治疗提供依据。
[1]孔令恩,吴倩仪,沈岩松,等.先天性肌强直一家系CLCN1基因突变的研究[J].临床神经病学杂志2012,25(06):407-409.
[2]Bandschapp O,Iaizzo PA.Pathophysiologic and anesthetic considerations for patients with myotonia congenita or periodic paralyses[J].Paediatr Anaesth,2013,23(9):824-833.
[3]Fialho D,Schorge S,Pucovska U,et al.Chloride channel myotonia:exon 8 hot-spot for dominant-negative interactions [J].Brain,2007,130(12):3265-3274.
[4]Statland J,Phillips L,Trivedi JR,et al.Muscle Channelopathies [J].Neurol Clin,2014,32(03):801-805.
[5]Ferriby D,Stojkovic T,Sternberg D,et al.A new case of autosomal dominant myotonia associated with the V1589M missense mutation in themuscle sodium channel gene and its phenotypic classification[J].Neuromuscul Disord,2006,16(5): 321-324.
[6]Portaro S,Altamura C,Licata N,et al.Clinical,Molecular,and Functional Characterization of CLCN1 Mutations in Three Families with Recessive Myotonia Congenita[J].Neuromolecular Med,2015,17(3):285-296.
[7]Tincheva S,Georgieva B,Todorov T,et al.Myotonia congenita type Becker in Bulgaria:Firs genetically proven cases and mutation screening of two presumable endemic regions[J].Neuromuscul Disord,2016,26(10):675-680.
R746.9 (
2017-01-09)
A
(责任编辑:李立)
10.3969/j.issn.1002-0152.2017.06.013
* 河北医科大学第一医院神经内科(唐山063000)△华北理工大学附属医院神经内科
猜你喜欢
世界科学技术-中医药现代化(2022年3期)2022-08-22 00:33:26
肝博士(2022年3期)2022-06-30 02:48:28
临床输血与检验(2022年3期)2022-06-22 02:52:50
世界科学技术-中医药现代化(2020年2期)2020-07-25 02:06:30
国际放射医学核医学杂志(2020年2期)2020-05-30 12:39:56
Journal of Sport and Health Science(2019年6期)2019-11-26 07:30:53
郑州大学学报(医学版)(2019年3期)2019-06-03 06:19:32
中国中医急症(2019年10期)2019-05-21 07:20:42
传染病信息(2019年2期)2019-05-17 13:16:04
现代检验医学杂志(2016年4期)2016-11-15 02:00:58
