
黑色素瘤转移相关基因的生物信息学分析
2017-08-16 04:21:56吴星袁定芬上海交通大学附属第六人民医院上海200233
中国中西医结合皮肤性病学杂志 2017年3期
吴星,袁定芬(上海交通大学附属第六人民医院,上海200233)
·论著·
黑色素瘤转移相关基因的生物信息学分析
吴星,袁定芬
(上海交通大学附属第六人民医院,上海200233)
目的 通过对与黑色素瘤转移相关的基因芯片进行生物信息学分析,为探索黑色素瘤转移的分子机制提供理论依据。方法 从公共基因数据库(GEO)中下载与黑色素瘤转移相关的基因芯片数据,经数据预处理后,分别通过BRB-Array Tools软件、DAVID在线分析软件、STRING在线数据库和Cytoscape软件进行差异表达基因的筛选、功能注释、通路分析、蛋白互作网络分析并计算网络及各个节点的拓扑特性。结果 BRB分析筛选出196个黑色素瘤转移差异表达基因,其中上调133个,下调36个,DAVID分析发现其主要集中在角质形成细胞分化、组织细胞间黏附、肥大细胞分化等生物学过程和Rap1信号通路、p53信号通路、谷胱甘肽代谢等通路。STRING分析发现了蛋白质网络互作图中的9个关键基因,分别为KRT14、KRT16、KRT1、EGFR、KIT、DSP、DSG1、PKP1、KLK7。结论 通过生物信息学的方法对黑色素瘤转移相关基因芯片数据进行2次挖掘,为进一步研究黑色素瘤转移的相关机制提供了理论依据。
黑色素瘤;生物信息学;基因芯片;BRBArray Tools软件
黑色素瘤是皮肤肿瘤中死亡率最高的恶性肿瘤之一,2010年全球黑色素瘤的死亡例数为46 372 例[1]。虽然黑色素瘤在中国的发病率较低,但由于我国人口基数巨大,黑色素瘤的发病在临床上也并不少见。转移是导致黑色素瘤预后不佳的重要因素,调查发现转移性黑色素瘤的中位生存期不足6个月,5年生存率不足5%[2]。
基因芯片,又名DNA芯片或DNA微阵列,具有高通量、高集成、微型化、自动化等特点,能够平行、快速地检测成千上万个基因转录体的表达水平,已广泛应用于突变基因检测、差异基因筛查、基因文库作图、药物作用靶标、肿瘤分型、多态性检测等多
个方面[3]。本研究通过对来源于基因芯片公共数据库(GEO)的黑色素瘤转移基因表达谱芯片数据进行分析,筛选出黑色素瘤转移差异表达基因,并对其进行GO富集分析和KEGG通路分析,同时作出其蛋白互相作用网络图,为进一步了解黑色素瘤转移的分子机制提供基础。
1 材料与方法
1.1 材料 本研究采用的黑色素瘤转移相关基因芯片数据(GSE8401)来源于美国国立生物信息技术中心(NCBI)的公共基因芯片数据库(GEO),芯片平台为GPL96[(HG-U133A)Affymetrix Human Genome U133AArray],其中原位黑色素瘤31例(GSM207929—GSM207959)、转移性黑色素瘤52例(GSM207960—GSM208011)。
1.2 方法
1.2.1 数据处理及差异基因筛选 利用BRB-Array Tools 4.5.1 Beta软件对从GEO中下载的黑色素瘤转移相关基因芯片数据集进行统计学分析。首先将数据按照Import data-Data importwizard的步骤导入软件,数据导入成功后,采用Just R MA算法和中位数法进行预处理、基因过滤及标准化,过滤的标准为:①两类样本的基因中位数至少发生3倍的改变,且这种改变不少于20%的样本数;②基因表达数据缺失数不超过50%;利用Class comparison工具对过滤标准的基因进行两独立样本t检验,找出原位黑色素瘤与转移性黑色素瘤的差异表达基因(P<0.001)。
1.2.2 差异基因生物信息学分析 利用DAVID在线分析软件(http://david.abcc.ncifcrf.gov/conversion.jsp)对黑色素瘤转移相关的差异表达基因进行GO富集分析和KEGG通路分析。
1.2.3 差异基因相互作用网络分析 利用STRING在线分析软件(http://string-db.org/)研究黑色素瘤转移相关的差异表达基因所编码的蛋白质之间的相互作用,绘制差异表达基因编码蛋白质的相互作用网络图。
2 结果
2.1 黑色素瘤转移差异表达基因筛选 根据预先设置的数据过滤标准以及中位数标准化处理后,BRB-Array Tools工具对原位黑色素瘤和转移性黑色素瘤两个样本进行分类比较,共同筛选出差异表达基因169个(表达差异3倍以上),其中上调133个,下调36个,上调和下调表达差异前10个基因见表1、2。

表1 上调差异表达基因 (TOP10)
2.2 上调差异表达基因生物信息学分析 通过DAVID在线分析软件对上调差异表达基因进行生物信息学分析。GO富集分析发现这些基因主要集中在表皮生长、角质形成细胞分化、信号传导、单个组织细胞间黏附、角化作用以及细胞骨架构成等生物学过程见表3。KEGG通路分析也发现其主要集中在Rap1信号通路、黑素合成、酪氨酸代谢、肾素分泌、p53信号通路、细胞间隙连接等通路,见表4。
2.3 下调差异表达基因生物信息学分析 通过DAVID在线分析软件对下调差异表达基因进行生物信息学分析。GO富集分析发现这些基因主要集中在肥大细胞分化、细胞间嗜同性黏附、交感神经分化、胚胎造血、对雌激素的应答、角质形成细胞分化等生物学过程见表5。KEGG通路分析也发现其主要集中在细胞外基质受体作用、脂肪酸降解、谷胱甘肽代谢等通路,见表6。
2.4 黑色素瘤转移差异表达基因相互作用网络分析 通过STRING在线数据库对169个黑色素瘤差异表达基因进行蛋白质相互作用网络分析并将其导入Cytoscape计算网络及各个节点的拓扑特性。
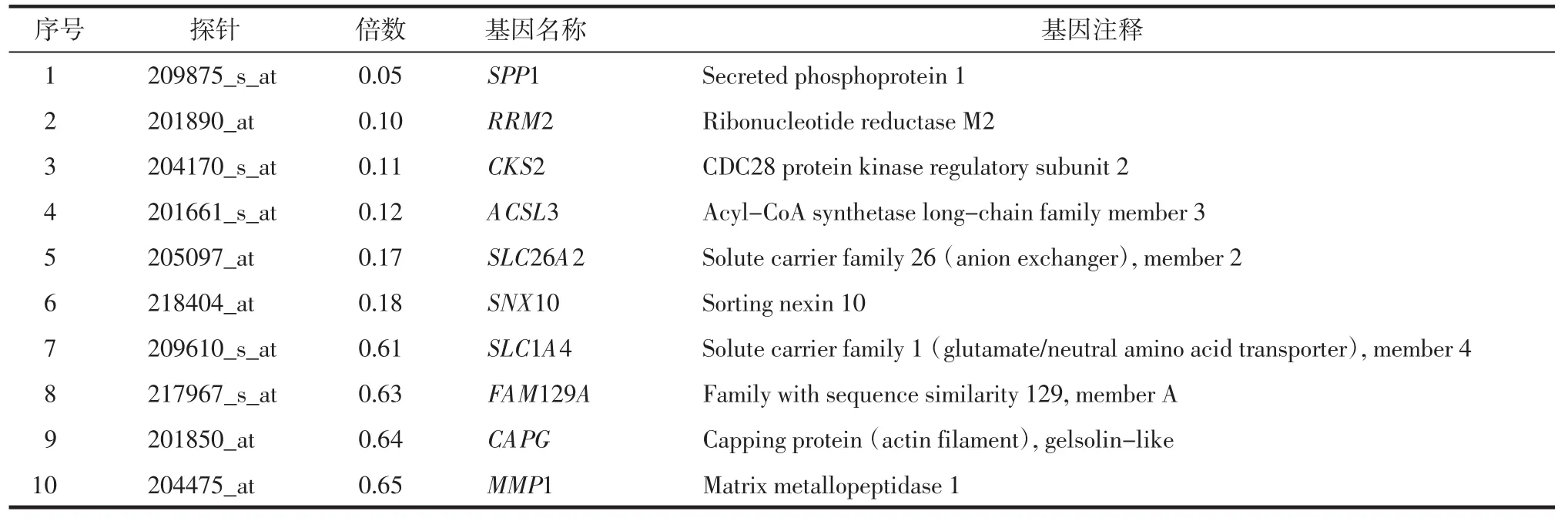
表2 下调差异表达基因 (TOP10)

表3 上调差异表达基因GO分析
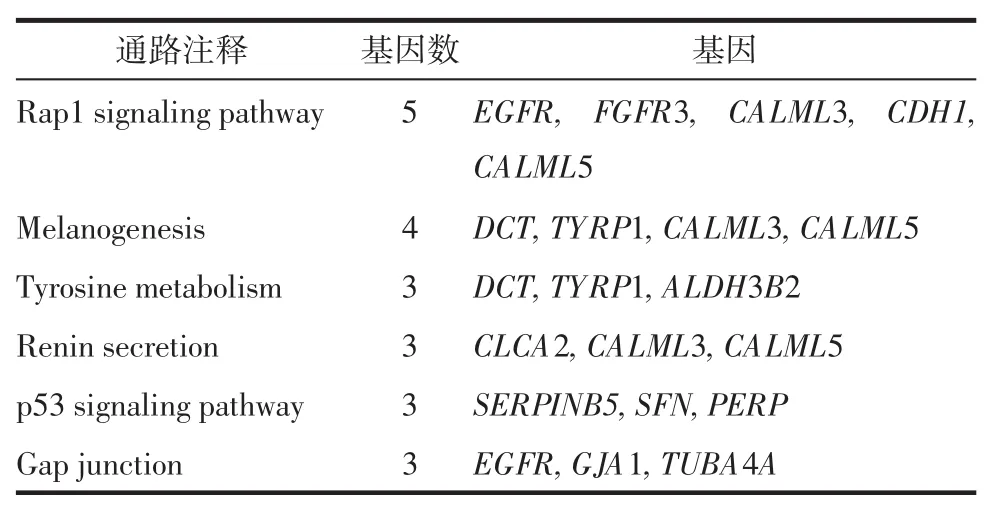
表4 上调差异表达基因KEGG分析

表5 下调差异表达基因GO分析

表6 下调差异表达基因KEGG分析
结果显示,125个基因编码的蛋白产物参与了蛋白质相互作用网络图的构建,其中大部分蛋白间都存在着相互作用,且有多个蛋白位于网络的中心,与周围蛋白关系密切。该蛋白质相互作用网络图由125个节点310条边组成,最大度值为16,最小度值为2,平均度值为(4.96±2.14)。本研究采用度数≥均数+ISD的蛋白所对应的基因为关键基因,他们共有9个,分别是KRT14、KRT16、KRT1、EGFR、KIT、DSP、DSG1、PKP1、KLK7见图1,其对维持网络结构的稳定至关重要。
3 讨论
黑色素瘤的死亡率长期以来都位居皮肤肿瘤的首位,而转移是导致黑色素瘤患者死亡的主要原因之一。目前黑色素瘤转移发生的具体机制尚未完全清楚,可能涉及多个方面,因此从分子水平揭示黑色素瘤转移的本质,研究黑色素瘤转移相关基因成为了黑色素瘤研究的热点之一。基因芯片技术是一种高通量、大量、快速、准确分析基因表达的工具,能够在不同条件、不同细胞类型、不同干预手段、不同细胞生长阶段及状态下对基因芯片做出相应的检测并产生海量的数据,使笔者能够运用生物信息学的方法对其进行整合分析,同时挖掘潜在的隐藏信息,从而为探索黑色素瘤转移的发生机制提供有利的线索。
本研究通过BRB-Array Tools软件对GEO数据库中的黑色素瘤转移相关基因芯片数据(GSE8401)进行分类比较,共发现差异表达基因169个,其中上调133个,下调36个。为了了解这些差异表达基因的生物学功能,笔者应用了DAVID在线软件对其进行了GO富集分析和KEGG通路分析,结果显示上调差异表达基因主要集中在集中在表皮生长、角质形成细胞分化、信号传导、单个组织细胞间黏附、角化作用、细胞骨架构成等生物学过程以及在Rap1信号通路、黑素合成、酪氨酸代谢、肾素分泌、p53信号通路、细胞间隙连接等通路,下调差异表基因主要集中在肥大细胞分化、细胞间嗜同性黏附、交感神经分化、胚胎造血、对雌激素的应答、角质形成细胞分化等生物学过程以及细胞外基质受体作用、脂肪酸降解、谷胱甘肽代谢等通路。该分析结果提示这些生物学过程和信号通路的变化可能在黑色素瘤的转移过程中发挥着重要的作用。对这些生物学过程及信号通路中涉及的基因进一步分析发现,他们之间存在着交集基因如EGFR、CDH1、CALML5、DCT、SPP1等,意味着这些基因可能在黑色素瘤的转移中起着协同作用,共同促进疾病的发生、发展。目前普遍认为黑色素瘤转移是一个多基因参与、多因素作用、多阶段发展的复杂过程,笔者的分析结果也证实这一观点,而这些异常的基因可能是未来治疗黑色素瘤转移的潜在靶点。

图1 差异表达基因蛋白质相互作用网络图
基于STRING在线数据库,笔者对黑色素瘤转移差异表达基因进行了蛋白质相互作用网络分析,发现125个基因蛋白产物参与了蛋白质相互作用网络图的构建,他们之间以线连接,且部分作为中心节点与周围的基因产物关系密切。本研究利用生物信息学工具筛选出了网络中的关键基因,他们是KRT14、KRT16、KRT1、EGFR、KIT、DSP、DSG1、PKP1、KLK7。其中,EGFR和KIT与黑色素瘤的研究在现有的文献报道中较多,而其他7个关键基因由于缺乏足够多的文献资料支持,有待以后更多的相关研究验证。
EGFR是一种具有酪氨酸激酶活性的跨膜糖蛋白受体,在黑色素瘤中MAPK和P13K/AKT信号通路是其下游的2个最主要的信号通路[4]。当EGFR被配体或自身突变磷酸化激活后,启动细胞内的信号传导,经过细胞质中的衔接蛋白和酶级联反应,导致MAPK信号通路持续活化以及P13K/AKT信号通路的激活,从而促进了黑色素瘤的增殖、分化、迁移、黏附以及血管生成,进而加速了黑色素瘤的转移。Huang等[5]发现在人黑色素瘤细胞系的裸鼠实验中EGFR的过表达与黑色素瘤的自发性转移存在一定的相关性。Slominski等[6]也发现EGFR在黑色素瘤中的表达增加会促进肿瘤的侵袭和转移。此外,Udart等[7]同样证实了EGFR活性随黑素瘤的发生发展及转移逐渐增强。以上均说明EGFR在黑色素瘤的转移中发挥了重要的作用,与本研究分析所得到的结果一致。
KIT是是Ⅲ型酪氨酸激酶受体家族中的成员,在其配体干细胞因子(SCF)激活KIT的胞外区域后,受体发生二聚化,经过特定的酪氨酸残端自身磷酸化或细胞内活化的酪氨酸激酶结构域,激活下游信号从而加速黑素细胞的形成、血细胞的形成、生殖细胞的发生以及肥大细胞的功能[8]。Monsel等[9]在黑色素瘤细胞系的实验中发现,KIT中K642E和L576P点突变会导致Ras/Raf/Mek/Erk信号通路的激活,从而刺激了黑色素瘤细胞的增殖和迁移。Minor等[10]则发现在黑色素瘤从原发部位到远处转移的不同阶段中,KIT的表达是逐渐下调的,表明这种下调可能致使黑色素瘤的预后不良。Kong等[11]同样发现了存在KIT基因突变的黑色素瘤患者中位生存期(30个月)远低于不存在此突变的患者的中位生存期(53个月)。这些都提示了KIT基因的突变与黑色素瘤的转移与预后紧密相连,对于疾病的预测以及预后的判断有一定的临床意义。
综上所述,本研究采用了生物信息学的方法对GEO数据库中的黑色素瘤转移相关基因芯片数据(GSE8401)进行了二次挖掘,成功筛选出196个差异表达基因,经GO功能注释和KEGG通路分析,为黑色素瘤转移的实验室研究提供了理论依据。此外,通过构建蛋白质互作网络图,得到了9个关键基因,其中EGFR和KIT的研究提示了其在黑色素瘤转移过程中的重要地位,而其他7个关键基因在黑色素瘤转移中的作用尚未明确,待人们进一步研究验证和挖掘。
[1] Jemal A,Bray F,Center MM,et al.Global cancer statistics[J].CA Cancer JClin,2011,61:69-90.
[2] 陈文静.恶性黑素瘤的治疗新进展 [J].医学综述,2013,19:54-56.
[3] Villasenor-Park J,Ortega-Loayza AG.Microarray technique, analysis,and appliance in dermatology[J].JInvest Dermatol,2013, 133:e7.
[4] Boone B,Jacobs K,Ferdinande L,et al.EGFR in melanoma: clinical significance and potential therapeutic target[J].JCutan Pathol,2011,38:492-502.
[5] Huang TS,Rauth S,DasGupta TK.Overexpressian of EGF receptor isassociated with spontaneousmetastasesofahumanmelanoma cell line in nudemice[J].Anticancer Res,1996,16:3557-3563.
[6] Slominski A,Ross J,Mihm MC.Cutaneousmelanima:pathology, relevant prognostic indicators and progression[J].Br Med Bull, 1995,51:548-569.
[7] Udart M,Utikal J,Krahn GM,et al.Chromosome 7 aneusomy.A marker for metastatic melanoma?Expression of the epidermal growth factor receptor gene and chromosome 7 aneusomy in nevi, primarymalignantmelanoma andmetastases[J].Neoplaia,2001,3: 245-254.
[8] Roskoski R Jr.Signaling by Kit protein-tyrosine kinase-the stem cell factor receptor[J].Biochem Biophys Res Commun,2005,337: 1-13.
[9] Monsel G,Ortonne N,Bagot M,et al.c-Kit mutants require hypoxia-inducible factor1 alpha to transform melanocyte[J].Oncogene,2010,29:227-236.
[10]Minor DR,Kashani-SabetM,GarridoM,etal.Sunitinib therapy for melanoma patientswith KITmutations[J].Clin Cancer Res,2012, 18:1457-1463.
[11]Kong Y,Si L,Zhu Y,etal.Large-scale analysisof KIT aberrations in Chinese patientswith melanoma[J].Clin Cancer Res,2011,17: 1684-1691.(收稿日期:2016-11-09)
·消息·
Bioinformatic Analysisof Differentially Expressed Genes in M etastatic M elanoma
Wu Xing,Yuan Dingfen
ShanghaiSixth People's Hospital,Shanghai Jiaotong University,Shanghai200233,China
Objective In order to provide theoretical base for themechanism of the developmentofmelanoma by screening differential genes related to this tumor based on bioinformatics analysis.M ethods In this research,we chose melanoma microarray datasets from GEO.After pre-processing the data,we used unpaired t-test to screen differential genes.We used tools in DAVID Software for GO analysis and KEGG pathway analysis,imported STRING online database for protein-protein interaction network analysis,and computed the network topology through Cytoscape software.Results We found 169 differentially expressed genes in melanoma in which 133 were up-regulated and 36 were down-regulated.GO analysis indicated thatmost of genes were enriched in keratinocyte differentiation,cell adhesion,mast cell differentiation,Rap1 signaling pathway and p53 signaling pathway.STRING software screened nine key genes including KRT14,KRT16,KRT1, EGFR,KIT,DSP,DSG1,PKP1 and KLK7.Conclusion The internalbiological information inmelanoma can be revealed by bioinformaticmethods,providing direction for further research inmetastaticmelanoma.
Melanoma;Bioinformatics;Microarray;BRBArray Tools
R739.5
:A
:1672-0709(2017)03-0197-05
猜你喜欢
现代畜牧科技(2021年4期)2021-07-21 06:12:50
今日农业(2021年4期)2021-06-09 06:59:56
中国博物馆(2018年2期)2018-12-05 05:28:50
西南国防医药(2016年6期)2016-12-01 06:01:13
现代检验医学杂志(2016年4期)2016-11-15 02:01:00
癌症进展(2016年10期)2016-03-20 13:15:41
哈尔滨医药(2015年3期)2015-12-01 03:57:44
实用手外科杂志(2015年4期)2015-08-27 01:54:26
现代检验医学杂志(2015年4期)2015-02-06 02:01:55
应用数学与计算数学学报(2014年2期)2014-09-26 05:40:23
