
纳米Fe3O4/超支化聚合物制备及性能
2017-08-16 10:26:03杨晓苏吴明华余德游
浙江理工大学学报(自然科学版) 2017年4期
杨晓苏,吴明华,余德游,田 丽
(浙江理工大学,a.先进纺织材料与制备技术教育部重点实验室;b.生态染整技术教育部工程研究中心,杭州 310018)
纳米Fe3O4/超支化聚合物制备及性能
杨晓苏a,吴明华b,余德游a,田 丽a
(浙江理工大学,a.先进纺织材料与制备技术教育部重点实验室;b.生态染整技术教育部工程研究中心,杭州 310018)
为了提高纳米Fe3O4的分散性,以马来酸酐改性超支化聚合物(简称超支化物)为模板,采用原位共沉淀法制备纳米Fe3O4/超支化物(Fe3O4/HB),并将Fe3O4/HB应用于催化双氧水降解染料。分析了铁盐比例(nFe2+∶nFe3+)、超支化物与FeCl2质量比(mHB∶mFeCl2)、吸附配位反应时间和共沉淀反应pH值对纳米Fe3O4粒径的影响,并对纳米Fe3O4/HB催化降解性能进行了测试。结果表明:纳米Fe3O4/HB制备的优化条件为:nFe2+∶nFe3+为1∶1.8,mHB∶mFeCl2为7.5∶1,吸附配位反应时间4 h,共沉淀反应pH值为11,所得纳米Fe3O4平均粒径为116.3 nm。Fe3O4/HB在中性条件下催化双氧水降解活性KN-G 60 min,其降解率可达到99.8%。相比于无超支化物为模板制备的纳米Fe3O4,实验所得纳米Fe3O4粒径小,分散性和催化降解性能明显提高。
超支化聚合物;纳米Fe3O4;粒径;催化降解
0 引 言
自Gao等[1]首次报道纳米 Fe3O4颗粒具有类似辣根过氧化物酶的催化活性以来,纳米Fe3O4仿酶催化剂被广泛运用于废水降解。制备纳米Fe3O4的方法主要有化学共沉淀法[2]、溶胶-凝胶法[3]、水热法[4]、乳液法[5]、高温分解法[6]、模板法[7]等。共沉淀法由于其制备过程简单、原料廉价易得而被广泛使用于Fe3O4颗粒制备[8]。由于纳米Fe3O4粒径较小、表面能较高且具有磁性,在制备过程和后续使用过程中容易团聚,影响纳米材料的应用性能。因此,如何提高纳米Fe3O4粒子的分散性是目前研究的热点[9-10]。超支化聚酰胺含有大量富电子官能团,能够捕捉溶液中金属离子,其内部的“空穴”为金属及其氧化物纳米材料的生成提供场所,对纳米粒子的生存起到很好的保护作用,从而防止纳米粒子的团聚,达到分散的目的[11]。
本文采用马来酸酐改性超支化聚酰胺为模板,借助模板分子上的双键和胺基与铁离子的配位作用,将金属离子吸附于超支化物[12],并通过添加碱剂调节pH使铁离子原位生成纳米铁氧化物,实现纳米Fe3O4在超支化物上的负载。本文对纳米Fe3O4/HB制备过程中影响纳米Fe3O4粒径的因素进行了研究,并对所得纳米Fe3O4/HB催化降解性能进行了测试。
1 材料与方法
1.1 实验材料与仪器
仪器:EL-300A型电子分析天平(常州市天之平仪器有限公司),DF-101S集热式恒温磁力搅拌器(河南省予华仪器有限公司),RE-52AA旋转蒸发仪(上海亚荣生化仪器有限公司),KQ-400KDB超声波清洗机(昆山市超声波仪器有限公司),Vertex7型傅里叶红外光谱仪(瑞士Bruker公司),LB-550型动态光散射纳米粒度仪(日本Honba公司),Lambda35型紫外分光光度计(珀金埃尔默仪器上海有限公司)。
1.2 实验方法
1.2.1 超支化聚酰胺的制备
超支化聚酰胺的制备方法参考文献[13],具体过程为:在氮气保护下,向250 mL四口烧瓶中加入0.2 mol二乙烯三胺,冰水浴冷却,再将0.2 mol丙烯酸甲酯与30 mL甲醇混合液,以恒压滴液漏斗缓慢滴入到烧瓶中。滴加完成后,室温下进行Micheal加成反应生成AB2型中间体。待反应4 h后,将反应液减压旋转除去甲醇;再加入0.01 mol马来酸酐,并缓慢升温到140 ℃,减压状态下,反应1 h后停止加热,自然冷却至室温。得到粘稠状的马来酸酐改性超支化聚酰胺(HB),合成反应方程式如图1所示。

图1 超支化聚酰胺合成路线
1.2.2 纳米Fe3O4/HB的制备
配制5 g/L的HB溶液150 mL,N2保护下,将按摩尔比1∶1.8称取的一定量的FeCl2·4H2O和FeCl3·6H2O 加入到HB溶液中,在多头磁力搅拌器上搅拌,使其吸附、配位4 h,然后在高速机械搅拌的条件下,使用恒压滴液漏斗缓慢滴加氨水调节pH值,升温至65 ℃水浴共沉淀反应90 min,再于80℃下静置陈化30 min,待反应完成后冷却至室温,用磁铁快速分离得到磁性颗粒。用无水乙醇反复洗涤颗粒,去除杂质,至pH值为7,将产物超声分散在无水乙醇中,备用,纳米Fe3O4/HB的制备如图2所示。
无超支化物为模板制备纳米 Fe3O4:用去离子水代替超支化物溶液,其它步骤同上制备纳米Fe3O4。

图2 纳米Fe3O4/HB的制备示意图
1.2.3 染料降解
在 50 mL浓度为 200 mg/L的活性翠兰 KN-G溶液中加入催化剂 40 mg/L,30% H2O28%,并调节 pH值为 7,控制温度 50 ℃,降解染料。每隔一段时间用紫外分光光度计在波长 664 nm处测降解液吸光度,根据以下公式计算其降解率。

其中:A0为反应开始时染料溶液的吸光度; At为降解一段时间染料溶液的吸光度。
1.3 测试方法
1.3.1 纳米Fe3O4/HB粒径的测试
将纳米Fe3O4分散液稀释100倍,采用LB-550型动态光散射纳米粒度仪在 25 ℃条件下测定其粒径。
将纳米Fe3O4/HB干燥样品置于Vertex7型红外光谱分析仪上,测定超支化(HB)和纳米Fe3O4/HB的官能团。采用ATR法测试,具体测定参数:分辨率为4cm-1,扫描频率为32s-1,波数为500~ 4500cm-1。
1.3.3 纳米Fe3O4/HB催化降解性能测定
以活性KN-G染料为降解材料,分析纳米Fe3O4/HB对双氧水的催化降解性能;在活性KN-G最大吸收波长(664nm)处,采用紫外分光光度计测定不同处理时间的染液吸光度,计算其降解率,评价纳米Fe3O4催化降解染料能力。
2 结果与讨论
2.1 纳米Fe3O4/HB制备工艺因素对纳米Fe3O4粒径的影响
2.1.1 氯化亚铁与氯化铁物质的量比的影响
制备Fe3O4时,理论需铁盐比 (nFe2+∶nFe3+)为1∶2,鉴于制备反应中Fe2+易被氧化成Fe3+,改变铁盐比,影响纳米Fe3O4的结构和磁性,从而影响纳米Fe3O4的催化降解染料的性能[14]。本实验在二价铁离子相对理论值过量条件下,研究铁盐比例对Fe3O4粒径的影响。
在nFe2+=0.5mmol,mHB∶mFeCl2=7.5∶1,吸附、配位反应时间为4h,共沉淀反应pH值为9的实验条件下,分别选取不同nFe2+∶nFe3+制备纳米Fe3O4/HB,测定纳米Fe3O4粒径,研究nFe2+∶nFe3+对纳米Fe3O4粒径的影响,结果如图3所示。
本次研究中甲状腺结节CNB患者84例,甲状腺结节87个,与术后病理诊断的结果相符率较高,这表明了,CNB对良性与恶性甲状腺结节的诊断精准性较高。本次成功穿刺86个结节,1个穿刺失败,是因结节较小,穿刺时取出的组织较少而导致失败,87个穿刺的结节中都没有发生过严重并发症,表明这种穿刺方式安全性较高。在恶性结节穿刺中,最大径小于1cm阶段就能确诊,这说明,在超声的引导下能够对恶性的甲状腺结节更早的进行诊断。

图3 nFe2+∶nFe3+对纳米Fe3O4粒径的影响
由图3可知,提高Fe3+用量的增大,纳米Fe3O4粒径逐渐减小。当nFe2+与nFe3+比例大于1∶1.2时,Fe3O4粒径减小不明显;nFe2+与nFe3+比例小于1∶1.2后,粒径减少速度加快;当nFe2+∶nFe3+为1∶2时,纳米Fe3O4粒径最小。增加nFe2+与nFe3+比例,可以抵消实验过程中部分被氧化的Fe2+。为了制备纯度较高、粒径较小的Fe3O4,为此实验选择nFe2+∶nFe3+为1∶1.8。
2.1.2 氯化亚铁与超支化物的质量比的影响
在nFe2+=0.5mmol,nFe2+∶nFe3+=1∶1.8,吸附、配位时间4h,沉淀反应pH=9的实验条件下,分别选取不同超支化物与氯化亚铁的质量比制备纳米Fe3O4/HB,测定纳米Fe3O4粒径,研究HB与FeCl2的质量比对Fe3O4粒径的影响,结果如图4所示。

图4 mHB∶mFeCl2对纳米Fe3O4粒径的影响
从图4可知,无HB时,Fe3O4的粒径为800nm左右;加入HB后,纳米Fe3O4的粒径急速减小;当HB与FeCl2的质量比超过7.5∶1后,粒径减少趋于缓慢。超支化物分子上含有大量氨基和不饱和双键,能与二价和三价铁离子吸附和配位,共沉淀反应时可以原位生成铁的氢氧化物或铁的氧化物,阻止了纳米Fe3O4团聚。HB的量越多,溶液中胺基和双键数量越多,与金属离子的吸附和配位几率越多,溶液中游离铁离子越少,生成的铁氧化物团聚越少[15];当质量比超过7.5∶1之后,体系内胺基和双键数量相对铁离子达到饱和或过量,此时再增加超支化物的量,对粒径的影响不是很大。
2.1.3 吸附、配位时间的影响
在nFe2+=0.5mmol,nFe2+∶nFe3+=1∶1.8,mHB∶mFeCl2=7.5∶1,共沉淀反应pH值为9的实验条件下,分别选取不同的吸附、配位时间制备纳米Fe3O4/HB,测定纳米Fe3O4粒径,研究吸附、配位时间对纳米Fe3O4粒径的影响,测试结果如图5所示。

图5 吸附、配位时间对纳米Fe3O4粒径的影响
由图5可知,当吸附、配位时间小于4h时,延长吸附、配位时间,纳米Fe3O4的粒径快速减小;当吸附、配位时间超过4h后,继续延长吸附和配位时间,纳米Fe3O4的粒径缓慢减小,并趋于平缓。其原因可能是延长吸附、配位时间,更多Fe2+和Fe3+被超支化聚酰胺的富电子基团诸如胺基和双键所吸附和配位,游离的Fe2+和Fe3+趋于减少,在共沉淀反应时原位生成的纳米Fe3O4的比例增多,游离Fe2+和Fe3+生成的纳米Fe3O4比例减少,因此纳米Fe3O4团聚几率小,所得的纳米Fe3O4粒径小。当时间超过8h后,超支化物上的富电子基团与金属离子的吸附和配位已趋完成,再延长时间,对纳米粒子的粒径影响不明显。
2.1.4 共沉淀反应pH值的影响


图6 pH值对Fe3O4粒径的影响
由图6可知,当pH小于11时,随着共沉淀反应pH值的增加,纳米Fe3O4的粒径逐渐减小;当pH值为11时,粒径达最小,为116.3nm;继续提高共沉淀反应pH值,粒径逐渐增加。其原因可能是氨水的加入促进铁离子盐转化为氢氧化物或脱水成氧化物。随着氨水的加入,所形成的氢氧化铁胶体表面所带的负电荷逐渐增多,胶体粒子之间产生电荷斥力,不易发生团聚[17];当pH值大于11后,共沉淀时pH值越高,形成氢氧化物沉淀速度过快,纳米粒子的团聚易发生,粒径就会变大。
2.2 纳米Fe3O4/HB红外光谱分析
图7为纳米Fe3O4/HB和HB的红外光谱图。HB的红外光谱图表明在1649cm-1处为C=O的伸缩振动峰,1260cm-1和1551.9cm-1处为C-N的伸缩振动峰和C-N的弯曲振动峰,属于HB上酰
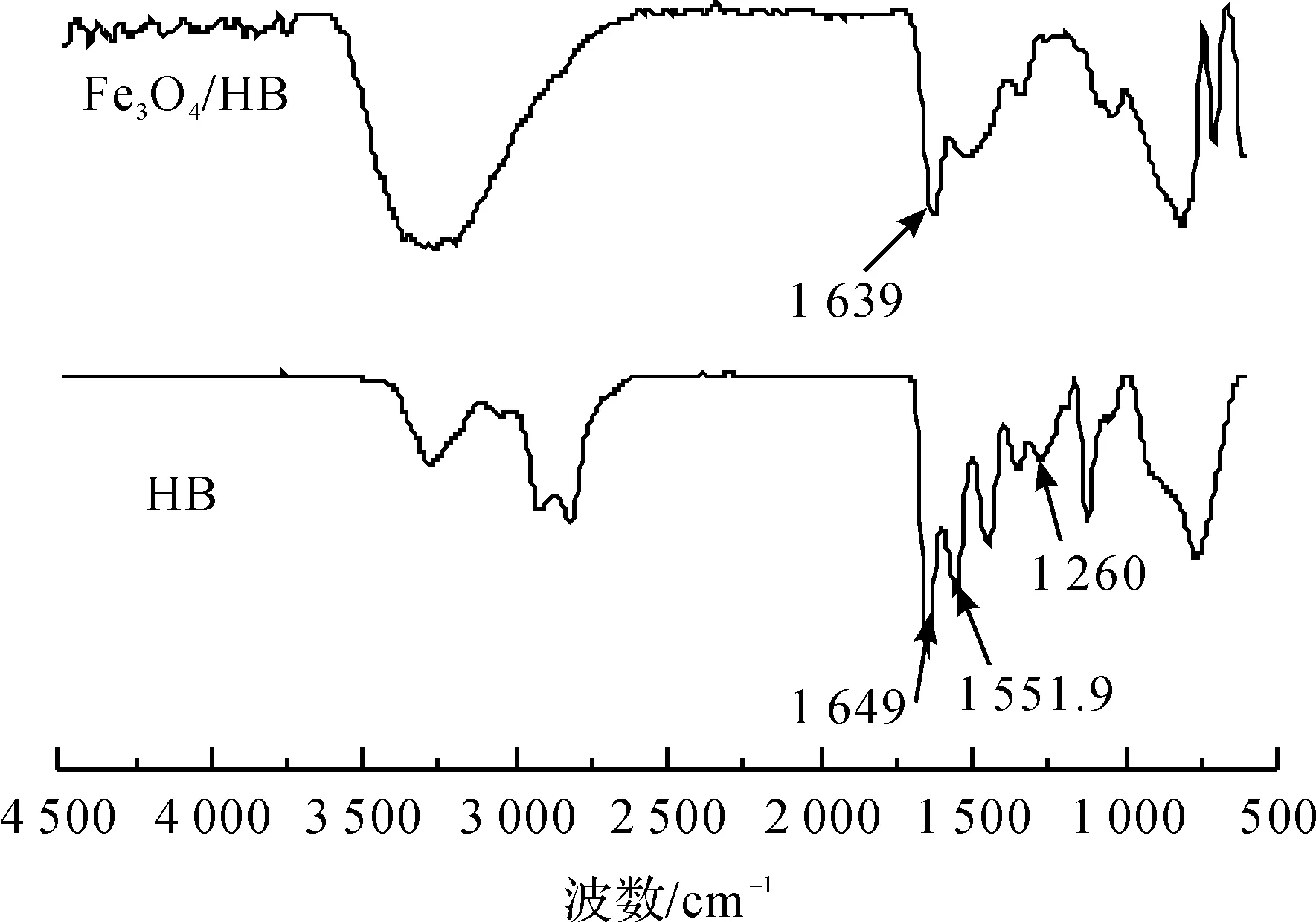
图7 红外光谱图
胺基团的特征峰。纳米Fe3O4/HB的红外光谱结果表明在1639cm-1左右出现了明显的吸收峰,为HB中的酰胺键的C=O伸缩振动峰,且该峰发生了位移。这说明实验成功制备纳米Fe3O4与HB的复合物。
2.3 纳米Fe3O4/HB的性能
2.3.1 纳米Fe3O4/HB的分散性
将纳米Fe3O4/HB和未加超支化物为模板制备的纳米Fe3O4经过磁分离、醇洗之后,再分散到乙醇溶液中配制分散液,观察其存放过程的稳定性,以此评判其分散性,结果见图8。图8中(a)为放置一天后纳米Fe3O4/HB分散液;(b)为放置一天后无超支化物为模板的纳米Fe3O4;(c)为放置三个月后纳米Fe3O4/HB分散液;(d)为放置三个月后无超支化物为模板的纳米Fe3O4。

图8 纳米Fe3O4分散液外观照片
从图8中(a)可以看出纳米Fe3O4/HB在乙醇溶液中具有很好的分散性,粒径分布在60~200nm之间(图9(a)),且放置三个月后纳米Fe3O4/HB仍处于良好的分散状态(图8(c));而图8 中(b)(无超支化物为模板制备的纳米Fe3O4)静置1d后在乙醇溶液中就会发生沉淀,且其粒径分布在500 ~850nm之间(图9(b)),放置三个月后沉降在底部的纳米Fe3O4团聚颗粒明显变大(图8(d))。造成纳米Fe3O4乙醇分散液分散性差别的原因主要是由于超支化聚酰胺是一种高度支化的大分子物质,其内部含有大量的氨基和双键,与金属离子之间形成吸附和络合作用,为纳米粒子的生成起到了很好的模板作用,防止了纳米粒子的团聚,使其粒径变小,不宜发生沉降[18];另外,在纳米Fe3O4分散液中,超支化物本身也起着良好的分散剂作用,分散稳定着纳米Fe3O4。

图9 纳米Fe3O4粒径分布
2.2.3 纳米Fe3O4/HB的催化性能
为了考察纳米Fe3O4/HB对双氧水的催化性能,比较了H2O2体系、Fe3O4H2O2体系和Fe3O4/HBH2O2体系对活性染料KN-G的降解率的影响。结果如图10所示。由图10可知,在反应前15min,纳米Fe3O4/HB降解染料效果相比于无超支化物为模板制备的纳米Fe3O4和不加任何催化剂提升不明显,这可能商品活性染料成分复杂,催化降解存在类似“诱导期”;但在反应30min后,Fe3O4/HBH2O2体系中降解率可达到86%;在反应60min时,纳米Fe3O4/HB催化H2O2降解KN-G,降解率可达到99.8%,而加入无超支化物为模板制备的纳米Fe3O4作为催化剂,催化效果不是很明显,仅35%,这说明Fe3O4/HB相比于无超支化为模板制备的纳米Fe3O4催化效果有很大的提升。

图10 不同催化剂对染料降解率的影响
3 结 论
a)以超支化物为模板采用原位共沉淀法制备纳米Fe3O4/HB可以明显减小纳米Fe3O4粒径,提高纳米Fe3O4分散性。纳米Fe3O4/HB最佳制备工艺条件为:nFe2+∶nFe3+为1∶1.8,HB与FeCl2质量比为7.5∶1,吸附、配位时间为4h,共沉淀反应时pH值为11。所得纳米Fe3O4粒径较小,为116.3nm左右;纳米Fe3O4乙醇分散液具有良好的稳定性。
b)在中性条件下,纳米Fe3O4/HB催化双氧水降解活性KN-G染料,降解率可达到99.8%,较未加超支化物为模板制备的纳米Fe3O4催化性能明显提高。
[1]GAOL,ZHUANGJ,NIEL,etal.Intrinsicperoxidase-likeactivityofferromagneticnanoparticles[J].NatureNanotechnology,2007,2(9):577-583.

[3]XUJ,YANGH,FUW,etal.Preparationandmagneticpropertiesofmagnetitenanoparticlesbysol-gelmethod[J].JournalofMagnetismandmagneticMaterials,2007,309(2):307-311.
[4]DENGH,LIX,PENGQ,etal.Monodispersemagneticsingle-crystalferritemicrospheres[J].AngewandteChemie,2005,117(18):2842-2845.
[5]FELTINN,PILENIMP.Newtechniqueforsynthesizingironferritemagneticnanosizedparticles[J].Langmuir,1997,13(15):3927-3933.
[6]LINF,CHENW,LIAOYH,etal.EffectiveapproachforthesynthesisofmonodispersemagneticnanocrystalsandM-Fe3O4(M=Ag,Au,Pt,Pd)heterostructures[J].NanoResearch,2011,4(12):1223-1232.
[7]BALOGHL,SWANSONDR,TOMALIAODA,etal.Dendrimer-silvercomplexesandnanocompositesasantimicrobialagents[J].NanoLetters,2001,1(1):18-21.
[8] 朱广谦,何青科,刘长庚,等.Fe3O4磁性纳米粒子的制备及其表面修饰[J].湖南师范大学自然科学学报,2016,39(3):46-55.
[9] 袁文俊,周勇敏.纳米颗粒团聚的原因及解决措施[J].材料导报:纳米与新材料专辑,2008,22(3):59-61.
[10]RUSEVOVAK,KOPINKEFD,GEORGIA.Nano-sizedmagneticironoxidesascatalystsforheterogeneousFenton-likereactions:InfluenceofFe(II)/Fe(III)ratiooncatalyticperformance[J].JournalofHazardousMaterials,2012,241:433-440.
[11]ZHANGD,ZHANGG,LIAOY,etal.SynthesisofZnOnanoparticlesinaqueoussolutionbyhyperbranchedpolymer[J].MaterialsLetters,2013,102:98-101.
[12]KAVITHAM,PARIDAMR,PRASADE,etal.GenerationofAgnanoparticlesbyPAMAMdendrimersandtheirsizedependenceontheaggregationbehaviorofdendrimers[J].MacromolecularChemistryandPhysics,2009,210(16):1310-1318.
[13] 朱鹏飞,吴明华,刘爱莲.马来酸酐改性超支化聚合物在棉织物纳米银整理中的应用[J].纺织学报,2016,37(1):104-109.
[14] 郑国华,陈洁,梁京祯,等.共沉淀法制备纳米Fe3O4的正交实验研究及特性[J].磁性材料及器件,2015,5:14-18.
[15] 杜宝吉,佘希林,杨光明.以树枝状大分子为模板制备纳米粒子研究进展[J].现代化工,2010,30(2):78-82.
[16] 陈亭汝,孙瑾.Fe3O4磁性纳米粒子的共沉淀法制备研究[J].应用化工,2009,38(2):226-228.
[17]CHENGFY,SUCH,YANGYS,etal.CharacterizationofaqueousdispersionsofFe3O4nanoparticlesandtheirbiomedicalapplications[J].Biomaterials,2005,26(7):729-738.
[18]MAHAPATRASS,KARAKN.Silvernanoparticleinhyperbranchedpolyamine:synthesis,characterizationandantibacterialactivity[J].MaterialsChemistryandPhysics,2008,112(3):1114-1119.
(责任编辑: 唐志荣)
Preparation and Properties of Fe3O4/Hyperbranched Polymer Nanoparticles
YANGXiaosua,WUMinghub,YUDeyoua,TIANLia
(a. Key Laboratory of Advanced Textile Materials and Manufacturing Technology, Ministry of Education; b. Engineering Research Center for Eco-Dyeing & Finishing of Textiles, Ministry of Education, Zhejiang Sci-Tech University, Hangzhou 310018, China)
In order to improve the dispersion of Fe3O4nanoparticles, Hyperbranched polymer modified by maleic anhydride was used as templates to prepare Fe3O4/Hyperbranched polymer nanoparticles (Fe3O4/HB) by a situ precipitation method. The nanoparticles were used to catalyze the degradation of dyes by hydrogen peroxide. The influence of the preparation factors including mole ratio ofnFe2+andnFe3+, the mass ratio of ferrous chloride and hyperbranched polymer, the adsorption and coordination time and precipitation reaction pH on the particle size of Fe3O4were explored. The catalytic degradation performance of the Fe3O4/HB was tested. The results showed that the Fe3O4nanoparticles exhibited small diameter around 116.3 nm, when the ratio ofnFe2+andnFe3+was 1∶1.8, the ratio ofmHBandmFeCl2was 7.5∶1, the coordination time was 4 h, the reaction pH was about 11. In nature conditions, the degradation rate of the KN-G dye could reach 99.8% in 60 min, when Fe3O4/hyperbranched polymer was used as catalyst. Compared with Fe3O4nanoparticles prepared without hyperbranched materials, the Fe3O4/HB with small particle size, dispensability and catalytic degradation performance were obviously improved.
Hyperbranched polymer; nanoparticles Fe3O4; particle size; degradation
10.3969/j.issn.1673-3851.2017.07.004
2016-10-12 网络出版日期: 2017-01-19
杨晓苏(1991-),女,湖北荆州人,硕士研究生,主要从事生态染整废水处理方面的研究。
吴明华,E-mail : wmh@zstu.edu.cn
TS190.3
A
1673- 3851 (2017) 04- 0491- 06
猜你喜欢
四川轻化工大学学报(自然科学版)(2021年1期)2021-06-09 06:12:08
建材发展导向(2021年24期)2021-02-12 02:00:02
中国粉体技术(2021年1期)2021-01-04 02:19:28
光谱学与光谱分析(2020年2期)2020-02-25 08:05:42
当代陕西(2019年6期)2019-04-17 05:04:10
山东青年(2018年2期)2018-06-23 11:17:58
中国塑料(2016年4期)2016-06-27 06:33:40
北方文学·下旬(2016年6期)2016-05-14 23:39:38
橡胶工业(2015年8期)2015-02-23 23:41:15
山东工业技术(2014年12期)2014-05-03 09:47:30
