
超分辨成像及超分辨关联显微技术研究进展∗
2017-08-08 06:57林丹樱屈军乐
物理学报 2017年14期
林丹樱 屈军乐
(深圳大学光电工程学院,光电子器件与系统教育部/广东省重点实验室,深圳 518060)
超分辨成像及超分辨关联显微技术研究进展∗
林丹樱 屈军乐†
(深圳大学光电工程学院,光电子器件与系统教育部/广东省重点实验室,深圳 518060)
(2017年4月19日收到;2017年6月21日收到修改稿)
光学成像系统中有限孔径对光波的衍射,使得光学显微成像技术的分辨率受到“衍射极限”限制而无法进一步提高.自1873年E.K.Abbe提出该问题以来,衍射极限就一直是学术界研究的热点.近年来,随着高强度激光、高灵敏探测器等光电器件研制技术以及新型荧光探针开发等相关领域的快速发展,光学显微技术衍射极限问题的研究迎来了新的契机,超分辨显微成像技术(super-resolution microscopy,SRM)在近十年内取得了令人瞩目的巨大成就.本文从空域和频域角度回顾了衍射极限分辨率的基本原理,并据此对目前常见的各种SRM技术“绕过”衍射极限提高分辨率的机理给予了详解,同时介绍了各类技术的发展动态和研究方向;作为SRM的一个新的重要的发展趋势,本文详细介绍了超分辨关联显微技术的最新研究进展,包括SRM与活细胞实时荧光显微、荧光寿命显微、光谱测量和成像、电子显微、原子力显微、质谱技术等的关联,着重讨论了各类超分辨关联显微技术的作用和意义;最后,对SRM技术和超分辨关联显微技术的未来发展方向进行了展望.
衍射极限,超分辨成像,关联显微技术,超分辨关联显微技术
1 引 言
光学成像系统中客观存在的有限孔径对光波的衍射使得光学显微镜的分辨率无法通过提高放大倍率和消除像差而无限提高,而是存在一个理论上限,称为衍射极限.该问题由E.K.Abbe于1873年提出显微镜的二次成像理论时指出并成为困扰光学设计界长达一个多世纪的难题[1],同时也一直是学术界研究的热点问题.近年来,随着高强度激光、高灵敏探测器等光电器件研制技术以及新型荧光探针开发等相关领域的快速发展,光学显微衍射极限问题的研究迎来了新契机,超分辨显微成像技术(super-resolution microscopy,SRM)在近十年内取得了令人瞩目的巨大成就,为现代生物医学研究提供了强有力的工具.2014年诺贝尔化学奖授予了E.Betzig,S.W.Hell和W.Moerner三位科学家,以表彰他们发明和发展SRM技术的贡献,其中前两位分别是单分子定位和受激辐射耗尽两类代表性SRM技术的发明人,第三位发现的荧光蛋白光激活效应则是单分子定位SRM技术的基础.他们的获奖肯定了化学、物理、生物等多学科交叉在当今前沿科学技术发展中的重要作用,同时也掀起了SRM技术和应用研究的新高潮.如今不仅SRM技术本身在快速发展和完善,SRM应用领域在迅速拓展和深化,而且SRM与其他技术还碰撞出了许多火花,例如超分辨关联显微技术就是近年来SRM的一个新的重要的发展趋势.本文介绍和分析各种SRM技术的基本原理和发展历程,并讨论将SRM技术与活细胞实时荧光显微、荧光寿命显微、光谱测量和成像、电子显微、原子力显微等技术进行关联显微的研究进展,同时对SRM技术和超分辨关联显微技术的未来发展进行展望.
2 光学显微镜的衍射极限分辨率
光学显微镜的衍射极限分辨率可从空域和频域两个角度来理解.
从空域角度,显微镜的成像过程满足透镜成像规律,理想情况下只要系统放大倍率足够高且很好地消除了各种像差,就能将任何极微小的样品放大到可被观察的程度.然而在19世纪,E.K.Abbe研究显微镜制造技术的改进时却发现显微镜的分辨率并不能无限提高,而且一个小孔径的消像差物镜甚至可能比一个大孔径的普通物镜得到更差的分辨率.从空域角度理解,其根本原因在于成像系统的有限通光孔径使光波发生衍射,导致物平面上无限小的物点在像平面变成有限尺寸的衍射斑,因此当两个物点相距太近导致两个衍射斑叠加形成一个像斑时就无法在图像上对它们进行区分了.也就是说,有限孔径的衍射使得光学成像系统存在一个分辨率上限,这就是衍射极限分辨率.显微镜中圆形孔径的衍射形成的像斑称为艾里斑[2],其原理可用图1(a)的单透镜成像来说明:物点O发出的光波经透镜L后在孔径A处发生衍射,波前各点发出的子波在像平面P相干叠加,几何像点P0处因各子波光程相同发生相长干涉而光强最强,P1点因光程相差半波长发生相消干涉而光强为零,其余则介于两者之间,故形成如图所示的艾里斑图样,其能量大部分集中在中央亮斑内,且孔径A越小、波长越长,P1离P0就越远,亮斑半径r越大.其定量关系为r=0.61λ/u′[3],其中λ为波长,u′为像方孔径角.根据系统观点,物点通过成像系统后形成的像斑称为点扩展函数(point spread function, PSF),系统的成像特性由该PSF决定.当两个物点同时经过系统时,其像斑将叠加,如图1(b)所示.可见,两个像斑存在一个临界距离,小于该距离时像斑叠加后将变得无法区分.该临界距离的确定通常采用瑞利判据,即以一个像斑的中心与另一个像斑的第一暗环重合作为区分标准,此时两像斑中心距离等于艾里斑半径.计算此时对应的两个物点的距离,可得到极限分辨率为d=0.61λ/NA,其中NA=nsinu称为数值孔径,n和u分别为物方折射率和孔径角.

图1 从空域和频域理解衍射极限分辨率 (a)系统孔径衍射形成的艾里斑;(b)区分两个像斑的瑞利判据;(c)频率分量、光学传递函数和截止频率Fig.1.Understanding di ff raction limit resolution from spatial and frequency domain:(a)Airy disk formed by system aperture di ff raction;(b)Rayleigh criterion for distinguishing two image spots;(c)frequency component, optical transfer function and cuto fffrequency.
从频域角度,显微镜的成像过程可用二次成像理论解释[1],即物体使光波产生衍射后,各级衍射波通过物镜聚焦到后焦面形成衍射图样,图样上各点发出的子波在像平面相干叠加形成像.其中物镜后焦面也称频谱面,其上形成的图样称为空间频谱,靠近光轴的频谱点对应小角度衍射波,距离光轴较远的频谱点对应大角度衍射波,每个频谱点的坐标对应一组空间频率.由此可推论,由于光学系统存在有限孔径,角度过大的衍射波将无法通过,从而频谱面上将不出现相应的频谱点,也没有相应的子波传播到像平面.也就是说,频谱面上的空间频谱将由于系统存在孔径而被限制在一个有限范围内,超出该范围的空间频率对应的衍射波将被滤除,可通过的最高空间频率称为系统的截止频率.对于荧光显微成像系统,只有平行于光轴的零级衍射波能全部通过,其他方向的衍射波随角度增大将逐渐被部分滤除.根据系统观点,该现象称为系统对不同频率成分有不同的传递能力,用光学传递函数(optical transfer function,OTF)表征,如图1(c)所示.系统的像方截止频率由孔径角u′和波长λ决定,定量关系为相应的物方截止频率为ξ0=2NA/λ.截止频率的存在,意味着得到的像缺少了来自物体的高阶衍射波的贡献,而这些高阶衍射波对应的正是物体的细节,系统无法传递这部分信息导致无法分辨与其对应的细节,因而分辨率存在上限.图1(c)模拟了三个截止频率不同的系统对同一物体的成像,其中代表物体光强分布的黑色曲线由图中蓝、红、绿三色代表的三个不同余弦分量叠加而成.可见,当截止频率较高、三个频率分量均能通过时,物体结构在像中得到了较好的重现;而当截止频率逐渐降低使部分高频分量不能通过时,得到的像将逐渐丢失相应细节.因此,截止频率决定了系统的分辨能力,其倒数即为极限分辨率,即d=1/ξ0=λ/2NA,这就是E.K.Abbe提出的极限分辨率公式,称为阿贝衍射极限.
3 超分辨显微成像技术原理及发展
3.1 超分辨的含义和实现途径
衍射是光波的基本性质,而显微成像系统的有限孔径又是客观存在的,因此如前所述的衍射极限分辨率,确实是一个客观存在、无法超越的理论极限.既然如此,又何以“超分辨”呢?实际上,近十几年来发展非常迅速的各种SRM技术,并非真的发明了不再受该理论极限限制的显微镜,而是通过各种巧妙的办法在衍射受限的系统中“绕过”衍射极限对图像分辨率的限制,获得分辨率高于衍射极限分辨率的超分辨显微图像.如果非要将从物体到超分辨图像之间看成一个系统的话,则该系统不仅包括荧光显微成像系统,还应包括获得超分辨图像所需的用来绕过衍射极限的手段.根据采取手段的不同,可将目前常见的各种SRM技术分为以下三类.
第一类是针对宽场成像系统的空域处理技术,以基于单分子定位的稀疏重构技术为主,代表性技术包括2006年几乎同时报道的三种技术,即Betzig等[5]发明的光激活定位显微(photoactivated localization microscopy,PALM),Rust等[6]提出的随机光学重建显微(stochastic optical reconstruction microscopy,STORM)和Hess等[7]报道的荧光激活定位显微( fl uorescence photoactivated localization microscopy,FPALM),其共同特点是控制荧光分子稀疏发光,使得一个衍射极限范围内基本不会有两个分子同时发光而导致不可区分,通过多次成像和定位获得各个荧光分子的位置而重构出超分辨图像.
第二类是针对扫描成像系统的空域处理技术,也称为点扩展函数工程(PSF engineering),以1994年Hell等[8,9]提出并于2000年实现的受激辐射耗尽(stimulated emission depletion,STED)技术为代表,其特点是针对点扫描系统中激光会聚于样品时因物镜孔径衍射形成一个艾里斑样激发斑的问题,利用套在激发光周围的高强度环形光使衍射极限范围内除中心点外的荧光分子发生受激辐射而不产生荧光,等效于将系统的PSF尺寸大幅缩小以获取超分辨图像.
第三类是频域处理技术,主要针对宽场成像系统,以2000年Gustafsson发明的结构光照明显微(structured illumination microscopy,SIM)[10]及其2005年在此基础上提出的饱和SIM(saturated SIM,SSIM)[11]为代表,其特点是从频域的角度提高分辨率,利用摩尔条纹将不可探测的高频信息转化为可探测的低频信息,即将超出截止频率以外的空间频率分量平移到截止频率范围内,因此也有人形象地称其为“移频法”.
3.2 针对宽场成像系统的空域处理技术
普通荧光显微( fl uorescence microscopy,FM)作为最简单的宽场荧光显微成像技术被广泛应用于生物医学领域.FM一般采用汞灯、氙灯或激光作为激发光源,对样品中的内源性荧光分子或外源性荧光探针进行全场激发,所发荧光由物镜收集后被面阵探测器(如电荷耦合器(charge-coupled device,CCD),互补金属氧化物半导体等)接收形成荧光图像.如前所述,由于系统孔径的衍射,样品中靠近的两个荧光分子形成的像斑中心距离若小于艾里斑半径则无法分辨.然而这里有个前提,即两个分子的光是同时被探测的,才导致两个像斑叠加无法区分.假如两个分子是先后发光并分别被探测的,则情况将大不相同.因为即使每次成像得到的仍然是艾里斑而不是理想像点,但由于已知每个像斑仅对应一个荧光分子,则此时不管像斑多大均可利用质心定位法、高斯拟合法等[12]确定其中心(理想像点)位置,从而将系统衍射的影响剔除;由于确定该点的过程不可能绝对精确,还原出来的像点尺寸将由定位精度(与探测到的光子数有关[13])决定.将两张图像做相同处理后再叠加在一起,此时只要还原后两个像点的半径小于其间距,叠加后将不会互相影响.这样一来,两个原本在分辨率极限内的分子就变得可分辨了,这就是基于单分子定位的稀疏重构技术“绕过”衍射极限实现超分辨的基本原理,如图2(a)所示.
然而,FM通常采用有机染料、量子点或荧光蛋白等作为荧光探针,标记或表达于样品中的特定分子或结构上,且一般需要足够高的标记密度方可如实反映被标记结构的特征,它们在宽场激发情况下将同时被激发而发射荧光,又如何能让靠近的两个荧光分子先后发光而不被同时探测呢?假如荧光分子具有开/关两个状态,即在相同激发条件下既有可发荧光的,也有不发荧光的,那么只要能控制每次探测时只有少数分子处于开态,则位于衍射极限内的两个分子同时发光的概率就会很小,这样每次探测得到的像斑对应一批单个的开态荧光分子,可通过定位获得它们的几何像点位置,然后通过反复的开关控制、探测和定位后将得到的大量像点叠加在一起,便可得到一幅分辨率远高于衍射极限的超分辨荧光显微图像.这就是1995年Betzig[14]提出的基于单分子信号实现超分辨成像的构思.该思路除了需要具有开关效应的荧光探针外,还需要探测器的灵敏度足够高、采集速度足够快,因此该方法能否实现,不仅依赖于荧光探针的开发和特性研究,也依赖于高灵敏探测器的发展.

图2 基于单分子定位的稀疏重构技术原理示意图 (a)PALM/STORM分辨衍射极限内两个荧光分子的基本原理;(b)利用柱透镜实现三维单分子定位的基本原理Fig.2.Schematic diagram of SRM technology based on single molecule localization and reconstruction:(a)Basic principle of resolving two fl uorescent molecules within the di ff raction limit by PALM/STORM;(b)basic principle of three-dimensional single molecule localization based on cylindrical lens.
2006年,Betzig等[5]利用光激活荧光蛋白(photoactivatable green fl uorescent protein,PAGFP)和高灵敏、低噪声的电子倍增CCD(electronmultiplying CCD,EMCCD)将其构思变成现实,实现了PALM技术.同时,Zhuang团队[6]和Hess等[7]也分别提出了基于相同原理的STORM和 FPALM技术,区别仅在于PALM和FPALM利用荧光蛋白的光激活效应[15]实现开关,而STORM则利用花菁染料Cy5的闪烁特性.这种基于单分子定位的稀疏重构SRM技术虽然只有短短十年左右的发展历史,但由于对成像设备要求不高,成为各类SRM技术中应用较广泛、研究最活跃的一类,近十年来发展非常迅速.以STORM为例,早期技术基于Cy3-Cy5染料分子对[6],Cy5的激发光在一定条件下将其由开态转至关态而不再发光,但采用短波长激光激活后可使关态分子重新转回开态而发光.这种转换与Cy3-Cy5分子对间的距离有关[16],经过巧妙设计的染料对在光漂白之前可被开关几百次,且每次处于开态时可探测到几千个荧光光子,定位精度可达10 nm左右.利用类似原理,Zhuang团队[17]进一步设计了不同的染料对并通过选择性激活实现了多色STORM.紧接着, Heilemann等[18]发现单独的Cy5(或Alexa 647)也可以直接用短波长激光激活实现开关,相应技术称为直接STORM(direct STORM,dSTORM),是目前普遍采用的形式.为提高纵向分辨率,研究者们又进一步在STORM等单分子定位SRM系统中引入柱透镜像散[19,20]或双螺旋点扩展函数等[21]方法,使轴向不同位置的分子形成不同形状或取向的像斑而可以区分和定位(如图2(b)所示),从而实现了三维STORM(3D-STORM).
然而,基于单分子定位的SRM也不全是依赖荧光分子开关特性的.例如,同样在2006年, Sharonov和Hochstrasser[22]提出利用溶液中自由扩散的荧光探针与目标间的瞬时结合和分离实现荧光分子稀疏探测的方法,称为点积累纳米成像(points accumulation for imaging in nanoscale topography,PAINT).该技术可采用普通荧光探针,样品制备简单且标记密度不受限制,但缺点是成像速度非常慢,获得一副超分辨图像往往需要几个小时甚至几天.此后发展的利用配体标记的uPAINT[23]和利用DNA标记的DNA-PAINT[24]或Exchange-PAINT等[25],对该技术进行了改进,但成像速度相比PALM和STORM等仍然慢得多,因此应用相对较少.2008年,Hell团队[26]则提出了先将大部分荧光分子激发至三重态,只探测剩余的或自发返回基态的少数分子的荧光以实现稀疏激发的基态损耗后返回技术(ground-state depletion followed by single-molecule return,GSDIM).由于原理相似,PAINT,GSDIM也经常与前面的PALM/FPALM,STORM一起被统称为单分子定位显微(single-molecule localization microscopy, SMLM).
实际上,要想将衍射极限范围内的多个荧光分子分开,除了不让它们同时发光外,只要还有其他手段能将叠加在一起的信号区分开,则即使它们同时发光也有可能实现超分辨.因此针对宽场成像系统的空域处理技术除SMLM外还有一些其他方法.例如,对于能够闪烁(包括自发间歇性发光)的荧光探针,由于每个荧光分子的闪烁过程是互不相关的,因此通过记录荧光分子的闪烁过程并对其随时间涨落的实时荧光信号进行高阶统计分析可区分不同分子信号,实现超分辨成像,这种技术称为超分辨光学涨落成像(super-resolution optical fl uctuation imaging,SOFI)[27].与SMLM不同的是,SOFI通常采用荧光量子效率更高的量子点进行标记,且不要求稀疏发光,成像速度更快.又如,荧光分子一般都是极性分子,当利用不同朝向的偏振光激发时,偏振方向与每个荧光分子的偶极矩夹角不同将导致荧光强度不同,通过周期性地改变偏振光方向并记录荧光强度随偏振方向的改变过程,也可以分析变化规律区分不同的分子实现超分辨,这种技术称为偏振反解调超分辨(super resolution by polarization demodulation,SPoD)[28].
近年来我国在这类宽场成像空域处理SRM技术方面的研究一直紧跟国际前沿,并取得了许多研究进展,例如中国科学院生物物理研究所的徐涛教授和徐平勇教授小组致力于研发新型光开关蛋白[29],华中科技大学的黄振立教授小组开发新的定位算法[30],张玉慧教授小组研究活细胞超分辨标记新策略[31],东南大学崔一平教授小组开展SMLM的生物应用研究[32],北京大学的席鹏教授小组报道了关于SOFI的工作[33,34],并与清华大学张奇伟教授小组等共同提出了一种新的基于SPoD的超分辨偶极子取向解析技术(superresolution dipole orientation mapping via polarization demodulation,SDOM)[35]等.深圳大学牛憨笨院士小组则针对3D-STORM对轴向漂移敏感的问题,设计了性能远高于市售设备的轴向防漂移系统,并结合3D-STORM和基于分子信标的荧光原位杂交技术实现了对长度仅2.5 kb的非重复基因组序列的三维超分辨成像,是目前国际上的最短记录[36].
3.3 针对扫描成像系统的空域处理技术
共聚焦激光扫描显微(confocal laser scanning microscopy,CLSM)是最典型的扫描荧光显微成像技术,它借助一对“共焦小孔”将焦平面以外的荧光信号滤除,不仅获得了远比普通FM清晰的图像,而且提高了纵向分辨率[37],具有光学层切和三维成像能力,在生物医学领域应用广泛.在扫描成像系统中,激光经物镜聚焦到样品上逐点扫描,扫描点上荧光分子的荧光由光电倍增管(photomultiplier tube,PMT)等点探测器收集,经数据处理形成一幅对应扫描点阵的图像,图像分辨率与扫描点阵有关.然而,是否缩小采样间距就能无限提高扫描成像系统的分辨率呢?答案是否定的.这是因为扫描成像系统同样受到衍射极限的限制,只是形式与宽场成像略有不同:宽场成像时衍射极限直观地表现为样品中一个荧光分子发出的光到达面探测器时变成了一个像斑,而在扫描成像系统中则更主要地体现在激光聚焦在样品上时不能形成一个理想的扫描点,而是形成一个艾里斑样的激发斑.因此每个扫描点处PMT收集到的荧光信号并非只来自理想扫描点,而是激发斑范围内全部荧光分子信号的累积,导致靠近的两个荧光分子在扫描得到的图像上同样无法区分.因此,对于扫描成像系统而言,提高成像分辨率的关键在于缩小激发斑尺寸,而该尺寸由衍射极限公式决定,无法无限缩小.
那么,在激发斑无法缩小的前提下,如何才能进一步提高成像分辨率呢?实际上这里的衍射极限限制仍然隐含着一个前提,即整个激发斑内的荧光分子同时发出荧光并被点探测器收集.假如可使激发斑内的荧光分子即使全部激发也只有中心点发荧光,则探测信号就只来自于理想扫描点,等效于将有效激发斑大幅缩小,提高成像分辨率.这就是Hell等[8,9]于1994年提出并在2000年实现的STED技术的基本思路,其令激发斑内中心点外其他分子不发荧光的办法,是利用一束与激发光共轴且波长与荧光分子发射波长匹配的环形光(称为STED光、损耗光或擦除光),使处于该环形光范围内的激发态分子由于受激辐射而迅速回落到基态,阻止它们发荧光,如图3(a)所示.扫描系统的激发斑也称激发PSF,与探测端的PSF共同决定荧光成像系统的PSF;对于一般的扫描成像系统,系统PSF更多地由激发PSF决定,因此Hell[38]也将其提出的STED技术称为PSF工程.在该技术中,环形STED光的产生和质量决定了等效激发PSF的尺寸,因此成为影响STED分辨率的关键.实验上产生STED环形光的方法一般是通过引入相位调制,使光束中心由于相消干涉而光强近似为零,例如在Hell最早的实验报道中采用的是在玻璃基板中心附近蒸镀一层光程为STED光半波长的氟化镁薄膜的方法,而目前常用的做法则是利用相位从0到2π连续变化的螺旋相位片来进行调制[39](图3(b)),使得调制后任意角度对称两侧的光波前均反相而在中心处相消干涉获得极小的暗点.而环形STED光能否有效地将激发态荧光分子通过受激辐射途径迅速耗尽,则与其功率密度密切相关,因此系统最终达到的分辨率由衍射极限分辨率和STED光功率密度I与饱和功率密度Is的比值共同决定,即d=λ/2NA·(1+I/Is)-1/2,其中Is为STED光将90%的激发态分子受激辐射耗尽所需的功率密度.可见,STED光功率越高,系统分辨率也越高,因此引入STED光后分辨率可以大幅提高到远高于衍射极限分辨率的水平(几十纳米甚至更高).
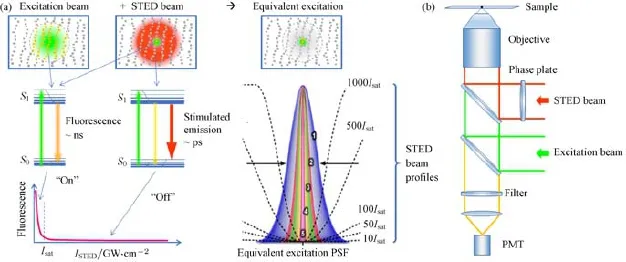
图3 STED技术原理示意图 (a)激发光、STED光、相应能级图和等效激发PSF;(b)STED光的产生及STED系统示意图Fig.3.Schematic diagram of STED:(a)Excitation beam,STED beam,corresponding energy level diagram and equivalent excitation PSF;(b)generation of STED beam and schematic diagram of STED system.
然而STED技术中Is一般为10—103MW/cm2量级,只有这样才能使激发态分子通过受激辐射返回基态的速率明显高于其通过自发辐射发出荧光返回基态的速率[40].这就意味着STED光必须是高强度脉冲激光,这不仅增加了系统造价,而且对样品损伤也较大,影响了STED技术在活细胞成像中的应用.因此,Hell[41]在提出STED思路后紧接着又提出了另一个设想,称为基态耗尽(ground-state depletion,GSD)技术,基本原理是将激发斑周边荧光分子抽运到三重态,使得只有扫描点附近的分子具备被激发至单重激发态并发出荧光的条件.许多荧光染料分子的激发过程本来就常常伴随这种从单重态到三重态的系间窜越过程(概率约为5%—20%),且三重态的亚稳态性质使得该过程更容易饱和,因此理论上利用该过程所需的饱和功率密度相比STED技术可低2—4个数量级,这一方面可使系统更容易获得更高的分辨率,另一方面可将损耗光强度大幅降低而使该技术更适合活细胞成像[40].Hell研究小组还进一步从理论上对STED和GSD技术进行分析,并将类似的抑制一部分荧光分子发光的途径归纳为可逆饱和线性荧光跃迁(reversible saturable optical fl uorescence transitions,RESOLFT)[40].2005年,他们利用具有开关效应的荧光探针在实验上实现了RESOLFT技术,并成功将损耗光功率密度降低了8个数量级[42].
目前国际上研究STED技术和应用的团队很多,而发明STED的Hell研究团队也在不断改进STED技术使其更适用于生物研究:GSD和RESOLFT等概念的提出和实现在降低对激光器要求的同时提高了该技术与生物样品的兼容性[43],双色STED技术的实现扩展了其应用范围[44],连续光激光器的采用降低了使用成本[45],快速STED技术实现了细胞囊泡运动、神经突触生长等活细胞动态过程的实时超分辨研究[46,47]等.在我国,北京大学的席鹏教授研究组[48]2012年首次在国内实现了STED技术,随后浙江大学的刘旭教授研究组[49]、中国科学院化学研究所的方晓红教授研究组[50]、北京大学的施可彬教授研究组等[51]也报道了STED方面的研究进展.针对光学系统像差和生物样品光学性质不均匀等导致STED对厚样品进行超分辨成像时分辨率急剧降低的问题,我们团队开展了利用相干光学自适应技术(coherent optical adaptive technique,COAT)改善STED光质量从而提高STED成像深度的研究工作,并取得很好效
果[52].
3.4 频域处理技术
如前所述,从频域角度,显微成像技术的衍射极限分辨率来源于系统OTF的截止频率,高于截止频率的成分被滤除,样品中周期小于截止频率倒数的余弦分量丢失.针对这一问题,Gustafsson[10]于2000年提出了SIM技术,基本原理是利用摩尔条纹把原本不能通过系统的高频信息平移到可观察的频率范围内来实现超分辨(图4(a)).具体地,是利用周期性结构照明光激发样品,使频域上由于结构光频谱与物频谱的卷积而产生携带物体信息的多级频谱.例如,采用正弦结构光可产生0级和±1级频谱,两两间距离ξ1为结构光周期的倒数,其中±1级频谱将携带物体细节信息的高频部分平移至截止频率范围内而被探测.但这些高频信息与0级频谱的低频信息叠加在一起(图4(b)),需要后期数据处理将三级频谱分开才能有效获得样品的高频信息.这种思路在信号处理或其他光学成像领域(如光学三维成像[53],X射线相衬成像等[54])也有广泛应用,且发展了较成熟的图像处理方法.SIM技术中通常采用相移法,基本思路是令结构光在一个周期内等间距平移若干次(≥3)并利用获得的多个叠加光强来求解.处理的结果使频谱范围从原来的(-ξ0,ξ0)拓展到(-ξ0-ξ1,ξ0+ξ1),因此ξ1越大(结构光周期越小)等效截止频率越高,分辨率越高.将结构光的取向进行旋转(图4(c))可实现二维平面的频率拓展.然而由于结构光条纹照射在样品上也受到系统衍射限制(ξ1≤ξ0),因此等效截止频率最多可拓展到2ξ0,即SIM的分辨率最多能在衍射极限基础上提高一倍.然而即便SIM的分辨率不如前两类SRM技术的高,由于不需要扫描且获取一幅超分辨图像需要的原始图像数量少,成像速度快,加上对样品制备和荧光探针没有任何特殊要求,对激发光功率要求也不高,光漂白和光损伤程度小,SIM在活细胞成像方面具有独特的优势.

图4 SIM技术原理示意图 (a)摩尔条纹的形成;(b)正弦结构光产生的移频效应;(c)二维空间频率拓展的实现Fig.4.Schematic diagram of SIM:(a)Formation of moiré fringes;(b)frequency shift produced by sinusoidal structured illumination;(c)realization of two-dimensional spatial frequency expansion.
光栅衍射告诉我们,正弦光栅只有0级和±1级频谱,而矩形光栅的频谱则要丰富得多,根据占空比的不同,除0级和±1级外,还可能出现±2级、±3级等其他高阶频谱.因此,假如能够产生一个占空比较小的矩形条纹作为结构光,则可以产生多级频谱,使等效截止频率得到多次拓展而大幅提高分辨率.但由于结构光的产生也受到光学系统衍射的限制,直接在物平面上形成一个周期接近衍射极限分辨率的矩形条纹并不现实.2005年, Gustafsson[11]在SIM的基础上提出了SSIM技术,巧妙地利用荧光分子的饱和激发使样品在正弦光波激发下发出具有高阶频率分量的非正弦分布结构荧光,从而实现多级频谱拓展,将分辨率提高到了几十纳米的水平.然而由于需要较高能量才能实现荧光分子的饱和激发,这种方法并不适合活细胞成像,无法发挥SIM的优势,因此并未被广泛应用. Heintzmann等[55]于2002年提出的饱和图案激发显微(saturated patterned excitation microscopy, SPEM)概念,也是基于类似的基本原理.
早期SIM系统采用置于显微镜管镜前焦面的透射式相位光栅来产生结构光照明[10],即令光栅产生的±1级衍射光分别会聚于物镜后焦面上对称的两点,从而在物平面上产生两束互成一定角度的光干涉形成结构光照明样品.通过调整物镜后焦面上两个会聚光斑的相对距离可改变结构光的周期,当两个会聚斑尽量靠近孔径边缘时得到频率最高(ξ1≈ξ0)的结构光.根据类似原理,Gustafsson团队[56]进一步通过改变光栅参数使物平面上产生三维结构光,将三维OTF的截止频率在横向和轴向上同时进行拓展,实现了三维SIM(3D-SIM).为克服原始图像获取过程中相位光栅平移和旋转对成像速度的限制,他们又引入空间光调制器(spatial light modulator,SLM)代替光栅,先后实现了二维和三维活细胞实时超分辨成像[57,58],并采用全内反射荧光(total internal re fl ection fl uorescence, TIRF)照明进一步提高成像质量.此后,他们又在光路中引入可调液晶相位延迟器实现了双色3D-SIM并用于活细胞研究[59].
不幸的是Gustafsson罹患癌症于2011年英年早逝,与其同在美国霍华德·休斯医学研究所Janelia研究园的PALM发明人Betzig承接了他的SIM实验室和研究小组并继续将SIM技术发扬光大.近几年来,Betzig团队致力于发展适合活细胞三维快速成像的光片显微(light sheet microscopy,LSM),先后发展了贝塞尔(Bessel)光[60]和格子(lattice)光片照明技术[61],并将其与SIM和PAINT等技术结合,实现了对厚样品的三维超分辨成像[62].2015年,Betzig等[63]将lattice-LSM和采用光控开关荧光探针的非线性SIM(nonlinear SIM,NL-SIM)结合在一起,不仅实现了对活细胞的双色三维快速成像,且空间分辨率最高可达45 nm,突破了SIM只能将分辨率提高一倍的限制,同时又避免了早期提出的SSIM中高功率激光对细胞的光毒性等问题,具有很好的发展前景.在我国,中国科学院西安光学精密机械研究所姚保利课题组[64]报道了基于DMD光调制的SIM技术.
3.5 三类方法的优缺点和发展趋势比较
综上所述,可以看出各类SRM技术虽然原理各不相同,但由于均是面向生物医学应用发明和发展起来的,因此不管对于哪种技术,提高分辨率均已不是惟一的追求,如何满足生物医学需要的多色、三维、活体等成像需求,一直以来也是各类SRM技术发展的目标.而由于三类方法各有优缺点,它们所面临的挑战也各不相同.
以活细胞成像为例:STORM和PALM等宽场成像空域处理技术,其分辨率取决于定位精度,可达到横向高于20 nm、轴向高于50 nm的水平,但由于重构一幅超分辨图像需要采集平均数万张原始图像,成像速度受限,通常只能用于固定细胞,尽管也有一些活细胞成像的报道[65],但到目前为止距离实际应用仍较远,这是因为活细胞应用的高帧频要求和单分子定位精度对光子数的要求是矛盾的,提高激光功率密度虽可一定程度上同时得到较高的帧频和光子数,但由此带来的光漂白问题限制了活细胞实时观察的时长(几十秒).因此,如何在较低功率密度和不降低光子数的同时提高帧频以适应活细胞实时超分辨成像的需求,还有赖于荧光探针闪烁特性的改善(包括探针结构的改进和化学环境的优化等)和活体标记技术的研究[66].
STED和RESOLFT等针对扫描系统的技术,其分辨率由擦除光功率密度与饱和光功率密度的比值决定,也可以达到几十纳米的水平,成像速度取决于扫描速度,与普通CLSM的成像速度基本一致,可以适应活细胞成像的需求,然而由于STED光功率密度相比STORM光功率密度要高4—6个数量级,因此对活细胞的光毒性和光损伤不能忽略,且高功率STED光在实现荧光擦除的同时也加剧了荧光分子的光漂白,这些都限制了STED在活细胞成像中的应用.采用光控开关荧光探针的RESOLFT技术一定程度上克服了这些问题,在活细胞成像方面具有一些优势;但由于原理不同,RESOLFT需要的光控开关荧光探针与现有的STORM探针不同,因为STORM需要探针单次闪烁期间光子数足够多,而RESOLFT则要求其在光漂白之前闪烁次数要足够多,因此,RESOLFT的进一步发展和应用,也与荧光探针的研发和闪烁机理研究有着密切关系.
频域处理的SIM技术由于仅需要采集九幅宽场图像便能恢复出一幅超分辨图像,因此不管是相比需要采集几万张原始图像的STORM/PALM技术还是相比需要点扫描的STED/RESOLFT技术,其成像速度都要快得多;而其激光功率密度比STORM/PALM技术还要低2—3个数量级,对样品的光漂白和光损伤等问题几乎可以忽略,因此在活细胞实时成像方面具有绝对的优势.然而由于成像原理的限制SIM只能将分辨率提高一倍,即最高100 nm左右;SSIM技术可以将分辨率提高到可与STORM/PALM或STED/RESOLFT相比拟的几十纳米水平,但饱和激发所需的高功率密度却使其失去了活细胞成像的优势;采用光控开光荧光探针的NL-SIM同时克服了这两方面的问题,在活细胞实时超分辨成像方面具有非常好的应用前景.
4 超分辨关联显微技术研究进展
超分辨关联显微技术是近年来SRM的一个新的发展趋势.关联显微的概念原指光学-电子关联显微(correlative light-electron microscopy, CLEM)[67],即将具有纳米分辨能力但特异性差的电子显微(electron microscopy,EM)和具有分子特异性识别能力但由于衍射限制只能分辨几百纳米的光学显微技术,尤其是FM相结合,从而架起微观世界和纳米世界之间的桥梁.广义地,关联显微也指利用具有不同特点的两种或多种显微成像技术同时或先后对同一样品的同一区域进行成像,并将获得的结果进行融合、对比和/或相关性分析,从而实现不同技术间的优势互补.
实际上,就像我们需要综合运用视觉、嗅觉、触觉等对一个物体的形状、颜色、气味、质地等做出判断方可对其全面认知一样,任何一种先进的显微成像技术都不可能单独提供研究一个问题的全面数据,甚至无法完备地描述一个体系的结构,因此这种优势互补在任何时候都是有益的.关联显微综合运用两种或多种先进显微成像技术,可以实现多维度、多尺度、多模态信息获取,从而综合得到生物样品在形态、功能、动力学、微环境和化学成分等多方面的信息[68].
近年来快速崛起的SRM技术为关联显微的发展带来了新的挑战和机遇.目前已有许多SRM与其他显微技术关联的报道.例如,2011年《Nature Methods》就报道了联用多种SRM技术和EM对多种蛋白在亚细胞水平进行定位的研究,获得了组蛋白H2B在细胞核中的高分辨定位和TOM20蛋白在线粒体中的定位[69].SRM缩小了普通FM与EM之间的分辨率差距,因此这种关联有助于提高结果相关性分析的准确性,使关联显微获得的结果更有意义.与此同时,SRM作为一种新兴技术,其与分辨率更高、技术相对成熟的EM技术的关联反过来也有助于对其所取得的新结果进行验证.除了SRM-EM关联显微,目前报道的基于SRM的关联显微技术还有很多,这些超分辨关联显微技术可以大致分为几类:SRM与其他衍射受限光学显微技术的关联,SRM与光谱测量技术的关联,以及SRM与非光学显微技术的关联.这些关联往往都能为同一个生物学问题提供不同尺度、不同维度或不同角度的信息互相印证,从而可使研究更加深入.
4.1 超分辨成像与其他衍射受限光学显微技术的关联
这一类型的关联是最直接的,因为两者有可能采用相同的样品制备流程和性质相似的荧光探针,也有可能基于相同或相似的光学系统进行成像,因而为关联带来了极大便利.这种关联的主要作用是弥补SRM目前的一些弱点,以便最大程度地发挥其在分辨率方面的优势,作用包括:识别和区分特定目标、提供多层次背景结构信息、提高时间分辨能力、丰富功能成像信息、实现多色超分辨等.
4.1.1 超分辨-普通荧光关联显微

图5 SRM-FM关联显微应用实例:识别和区分特定目标[70]Fig.5.Correlative SRM-FM applications:Identifying and distinguishing speci fi c targets[70].
SRM通常采用高倍物镜进行成像,其视场一般仅有几十微米,直接在SRM成像模式下寻找特定目标往往比较困难.而传统FM作为最简单、应用最广泛的荧光显微成像技术,在特定细胞或结构的标记和判别方面已形成了一套比较完整的体系.因此,将SRM与普通FM进行关联,可以先利用FM在较低分辨率下快速寻找到感兴趣区域后再切换到SRM模式进行超分辨成像,还可以采用与SRM成像所用荧光探针具有不同光谱性质的其他探针对特定细胞或结构进行特异性标记,以更好地识别和区分感兴趣的目标.例如,Xu等[70]利用3D-STORM技术对神经元细胞骨架进行超分辨成像研究的工作中就采用了这种SRM-FM关联技术来区分神经元中的树突和轴突两种不同的突触结构.如图5所示,他们除了使用联接有荧光分子Alexa 647的鬼笔环肽标记肌动蛋白微丝外,还利用另一种荧光探针Alexa 555免疫标记了树突中的特异性蛋白MAP2(图5(a)和图5(d))和轴突起始段中的特异性蛋白NrCAM(图5(f)),首先利用传统FM成像区分树突和轴突,再利用3D-STORM对两者的微丝结构进行SRM成像(图5(b),(e)和(g)),从而发现神经元两种突触中的微丝具有完全不同的超分辨结构,其中轴突的微丝具有明显的周期性结构.
4.1.2 超分辨-共聚焦关联显微
目前,虽然如前所述各种SRM都在努力实现多色成像,但多数SRM技术由于对荧光探针或光学系统有特殊要求,还不能方便地实现很好的多色成像.而CLSM作为另一种在生物医学领域应用广泛的荧光显微成像技术,其多通道成像功能可以很好地弥补SRM这方面的不足:在利用SRM对一些细微结构进行超分辨成像之前,可先通过CLSM获取多色标记样品的背景结构信息,这样一方面起到与SRM-FM关联显微相同的识别和区分特定目标的作用,另一方面由于CLSM可以提供比普通FM更清晰的不同尺度的图像,尤其是提供组织、细胞、亚细胞等不同层次的结构信息作为分析超分辨结构的背景,从而有助于分析超分辨图像的生物学意义.例如,Crossman等[71]报道了一种用于多尺度组织成像的dSTORM-CLSM关联显微技术,他们首先对人心脏组织中含有心肌亲联蛋白JPH(Alexa 680标记)和兰尼碱体RyR(Alexa 750标记)的大分子膜复合体进行超分辨成像(图6A(c)),接着利用CLSM对这两种蛋白以及标记了麦胚凝集素WGA(Alexa 594标记)的细胞膜进行成像(图6A(a)和(b)),通过融合分析两种方法获得的图像,将dSTORM结果分割到细胞膜和细胞质两种不同区域进行统计分析(图6A(c),(d)和(e)),从而发现RyR与JPH在质膜上有更高的共定位水平.Barna等[72]采用类似的方法对固定的心脏和肾脏组织切片等进行了STORM-CLSM关联显微成像(图6B),并详细报道了利用开源软件VividSTORM对该类型关联显微图像进行分析以获取特定细胞或亚细胞结构内某种分子的含量、密度、间距等定量信息的流程.
4.1.3 超分辨-实时荧光关联显微
虽然目前各种SRM技术都有不少关于提高成像速度的报道,其中甚至不乏帧频达到视频级别的结果[46,73],然而在实际应用中多数SRM技术要想将获得单幅超分辨图像的时间控制在10 s以内仍然是比较困难的,尤其是对分辨率和图像质量要求较高时,而这对于活细胞中的一些动态过程而言显然还是太慢.因此将具有高空间分辨能力的SRM和空间分辨率相对较低但时间分辨率可以很高的活细胞实时荧光显微成像(live-cell timelapse fl uorescence microscopy,LTFM)[74]相关联,是目前提高SRM时间分辨能力的一个有效途径. Bálint等[75]将用于活细胞的LTFM与用于固定细胞的3D-STORM相关联,结合单粒子追踪(single particle tracking,SPT)技术,通过追踪活细胞中单个溶酶体的运输路径并与微管的超分辨结构进行关联分析(图7),研究了溶酶体在微管交接处的行为特征,从而为马达蛋白驱动的囊泡运输机理研究提供了丰富的数据和新的视角.这种关联的难点之一是要确保超分辨成像时固定细胞的骨架结构与活细胞成像阶段的骨架结构一样,而该工作的巧妙之处就是在活细胞成像阶段对细胞采用药物处理使细胞既能保持活性又能使骨架保持基本不变, LTFM成像结束时再原位对细胞进行固定和免疫荧光标记等一系列超分辨样品处理流程并进行超分辨成像,为此该研究小组还发明了一种能够原位实现细胞固定和超分辨标记的装置[76],采用计算机控制样品处理过程中所需的各种不同试剂的自动注入.

图6 SRM-CLSM关联显微应用实例:提供多层次背景结构信息[71,72]Fig.6.Correlative SRM-CLSM applications:providing multi-level background structural information[71,72].
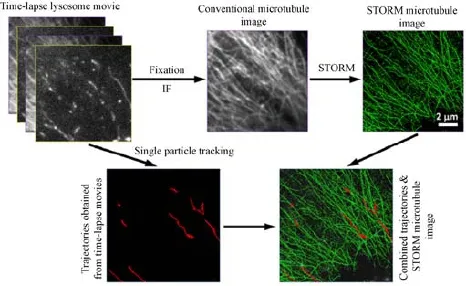
图7 SRM-LTFM关联显微应用实例:提高时间分辨能力[75]Fig.7.Correlative SRM-LTFM applications:improving time resolution[75].
4.1.4 超分辨-荧光寿命关联显微
荧光寿命显微成像( fl uorescence lifetime imaging microscopy,FLIM)技术通过探测样品中荧光探针的荧光衰减速率来反映其与微环境的相互作用,已被广泛应用于定量检测细胞中的离子浓度、pH值、氧含量、黏滞度、折射率等多种生物化学或生物物理参数,是一种典型的功能成像技术.而目前多数SRM技术主要是针对结构进行成像,因此SRM-FLIM关联显微可为SRM技术提供一个正交维度,丰富其功能成像信息.例如,Auksorius等[77]报道的STED-FLIM技术,首次获得了高于衍射极限分辨率的FLIM图像.另一方面,利用不同荧光分子荧光寿命的不同,可以区分光谱性质相似的荧光探针,将这一性质应用于SRM技术还有助于实现多色SRM.例如,Hell研究团队[78]利用STED-FLIM关联实现了双色STED技术,他们利用荧光探针ATTO 647N和KK 114分别标记了固定细胞中的微管蛋白和核纤层蛋白并进行STED成像,同时在扫描过程中利用时间相关单光子计数技术(time-correlated single-photon counting,TCSPC)对荧光分子的寿命进行测量,由于两种荧光分子的吸收光谱和发射光谱均很相似,系统只需采用一束激发光和一束STED光,而获得的荧光强度图像中两种分子的荧光信号将重叠在一起(图8(a)),但由于两种分子的荧光寿命明显不同,因而可利用寿命信息将两个通道分开(图8(b)),从而实现了单激发波长的双色STED.此外,我们团队还将SMLM与FLIM进行关联,利用单分子定位算法和反馈控制发展一种能够对活细胞中的运动目标进行荧光寿命追踪的方法[79],进一步与SMLM获取的超分辨结构结合将有助于揭示细胞中微环境变化或相互作用与超分辨结构间的关系.

图8 SRM-FLIM关联显微应用实例:实现多色超分辨[78]Fig.8.Correlative SRM-FLIM applications:Realizing multi-color super-resolution[78].
4.2 超分辨成像与光谱测量技术的关联
光谱测量可以提供样品的化学成分或分子结构信息.然而多数光谱测量技术是针对溶液、粉末等样品的,获得的往往是样品中分子的统计信息,而不包含样品的空间分布信息,也不能直接对单个分子的光谱进行测量.基于光谱测量的光谱成像技术虽可利用样品不同位置的光谱信息进行成像,但分辨率一般较低.将SRM技术与光谱测量技术相关联,可大幅提高光谱成像技术的空间分辨率,同时也为SRM增加额外的信息维度.
4.2.1 超分辨-荧光光谱关联显微
由于SRM技术本身就是基于探测荧光的,因此将其与探测荧光发射光谱的荧光光谱测量技术( fl uorescence spectrometry,FS)进行关联是比较直接的一种关联方式.这种SRM-FS关联显微一方面便于实现单分子FS测量和成像,另一方面还可为SRM技术提供实现多色成像的另一个途径.Zhang等[80]利用STORM-FS关联同时实现了高通量单分子FS测量和光谱分辨的多色STORM技术.如图9A所示,他们搭建了一套双物镜系统(图9A(a)),其中一个物镜用于获取单个荧光分子的位置信息(图9A(b)),另一个物镜采集的单分子荧光信号经过一个棱镜后产生的色散被同时记录下来(图9A(c)和(d)),这样单次曝光即可同时获得上百个单分子荧光光谱,并且由于STORM中荧光分子的稀疏激发,这些单分子荧光光谱大多不会互相重叠,从而实现了每分钟106个的高通量单分子FS测量.通过对STORM图像中每个荧光分子的光谱进行分析,还可以获得每个分子的发射峰值波长并据此赋予相应的颜色,从而形成一幅具有“真彩色”的超分辨图像,实现光谱分辨的多色STORM技术.如图9B所示,他们采用了四种发射中心波长彼此仅相差10 nm左右的荧光分子对细胞中四种不同结构同时进行标记,利用这种光谱分辨STORM技术可以很好地区分四个通道,串扰率小于1%.随后,Hess研究团队[81]又报道了一种基于单物镜的FPALM-FS关联技术,他们采用分束器将物镜收集的荧光分为两部分,分别用于单分子定位和单分子FS测量,虽然这样一来不可避免地减少了各光路中的光子数,但系统设计相对简单,且单物镜系统对样品的要求比双物镜系统要低很多,更适合细胞成像.
4.2.2 超分辨-红外光谱关联显微
相比FS测量和FM成像,红外光谱技术的优点在于无需标记,可提供样品的化学信息.近年来,同步辐射傅里叶变换红外(synchrotron radiation-based Fourier transform infrared spectromicroscopy,SR-FTIR)光谱显微的最新研究进展已揭示了细胞中一些很有价值的化学信息[82],然而其空间分辨率仅为5µm左右.Whelan等[83]报道了将SR-FTIR光谱测量技术和SMLM进行关联并用于单细胞研究的工作,他们对固定前后的细胞进行SR-FTIR光谱分析(图10(a)—(c)),然后再对免疫荧光标记后的同一个细胞的细胞骨架微管蛋白进行超分辨成像(图10(d)—(k)),通过对两种结果进行相关性分析,观察到采用不同固定方法造成的细胞不同程度的成分和结构变化,从而为优化超分辨样品制备流程提供了一种新的手段.
4.3 超分辨成像与非光学显微技术的关联
由于图像衬度形成机理的不同,SRM与EM等非光学显微技术对样品制备的要求往往很不相同,甚至有可能互相冲突,这给两者关联带来了极大的挑战,成为实现这类关联必须解决的关键技术问题.近20年来,关联了普通FM与EM的CLEM技术已取得了丰硕的研究成果[84-86],关联样品制备技术也日趋成熟.因此在普通FM基础上发展起来的SRM技术与EM的关联相比SRM与其他非光学显微技术的关联具有更坚实的基础,其成果也更丰富.
4.3.1 超分辨-电子关联显微

图9 SRM-FS关联显微应用实例:实现高通量单分子FS测量和光谱分辨多色超分辨[80]Fig.9.Correlative SRM-FS applications:Realizing high throughput single molecule FS measurements and spectrally resolved multi-color super-resolution[80].

图10 SRM-IR关联显微应用实例:架起结构变化和化学成分变化之间的桥梁[83]Fig.10.Correlative SRM-IR applications:Bridging between structural changes and chemical composition variations[83].
EM技术包括扫描电子显微(scanning electron microscopy,SEM)和透射电子显微(transmission electron microscopy,TEM)两类,前者对样品表面成像,分辨率高于10 nm;后者对超薄切片(约50—100 nm)成像,分辨率可达亚纳米.EM成像过程需要在真空环境下进行,对样品制备有严格要求,对生物样品而言通常需要固定、脱水、干燥、包埋、切片等环节,此外SEM还需要导电处理,TEM一般还需要染色.其中染色是因为生物样品中主要为轻元素原子,要形成较强的图像衬度需要利用重金属对脂类或蛋白质进行染色,但这种染色并非特异性标记,因此EM的分子特异性识别功能很差.利用联接有金纳米颗粒的抗体对样品进行免疫标记可提供较好的分子特异性,但目前其应用还受到标记密度低、可标记目标分子少等限制.将SRM与EM进行关联可弥补EM在分子特异性识别方面的缺点,实现优势互补;但EM样品制备流程中脱水、干燥等环节对于SRM样品中的荧光探针有着致命的影响,因此SRM和EM不仅不能同时成像,而且样品制备和成像流程都必须经过优化设计才有可能实现二者的有效关联.SRM-EM关联最早在Betzig等[5]提出PALM技术的同时就开始了,他们采用CLEM中应用较多的冷冻切片TEM样品制备技术,利用PALM-TEM关联结果的高度相关性证明了利用PALM技术直接对细胞内蛋白质进行超分辨成像的有效性和准确性(图11).随后其他SRM-EM关联技术也陆续发展起来,关联样品除冷冻切片还有树脂包埋切片、非切片等,而针对不同样品需要不同的处理和不同的成像流程[87].以SRM-SEM关联为例,Löschberger等[88]利用dSTORM-SEM关联研究了非洲爪蟾卵母细胞核膜中的核孔复合物,他们首先对分离出来的核膜进行常规的dSTORM样品制备并进行超分辨成像,接着用较高浓度的戊二醛进行后固定并依次进行四氧化锇固定、丙酮脱水、临界点干燥等处理,最后覆盖一层碳以提高样品导电性用于SEM成像.通过分析dSTORM-SEM关联结果,他们发现大多数NPC由8个核膜糖蛋白gp210的同源二聚体(图12(e)—(h))组成(少数含有9个).最近,Wojcik等[89]提出了利用石墨烯直接对湿样品进行STORM-SEM关联显微的新方法.厚度极薄(单原子层)的石墨烯膜具有可穿透光和电子、可完全隔绝气体和液体、可导电和不易发生化学反应等优点,因此在细胞样品表面覆盖一层石墨烯膜则无需对细胞进行后固定、脱水、干燥、镀导电膜等处理,不仅大幅简化了样品制备流程,而且降低了对细胞固定和荧光标记的要求,将使SRM-EM的发展前景更加光明.

图11 SRM-TEM关联显微应用实例:验证超分辨成像结果[5]Fig.11.Correlative SRM-TEM applications:Validating super-resolution imaging results[5].

图12 SRM-SEM关联显微应用实例:实现分子特异性超分辨[88]Fig.12.Correlative SRM-SEM applications:Realizing molecular speci fi city in super-resolution imaging[88].
4.3.2 超分辨-原子力关联显微
原子力显微(atomic force microscopy,AFM)作为另一种常用的表面成像技术,广泛用于纳米技术和材料领域,通过测量探针与样品表面的相互作用实现对样品表面形貌的三维成像,横向分辨率为10 nm左右,纵向分辨率可达亚纳米量级.此外,利用AFM还可以进行作用力和弹性模量等力学参数的测量;换上专用的金属涂层光纤探针则可实现近场扫描光学显微(near- field scanning optical microscopy,NSOM)功能,即早期 Betzig和Trautman[90]提出的近场超分辨技术.由于AFM成像无需真空环境,对样品制备要求不高,因此近几年也越来越多地被用于生物样品成像,甚至可对活细胞或组织直接进行成像[91,92].因此SRMAFM关联相比SRM-EM要简单得多.例如,Harke等[93]利用可安装在倒置显微镜上的AFM扫描装置实现了STED-AFM关联,并通过荧光珠进行了实验验证.此后这种关联方式也被广泛应用于其他SRM与AFM的关联[94,95].不过,由于SRM采用的油浸物镜会受到AFM悬臂振动的影响,且两者采用的激光会彼此干扰,所以即使是集成于一套系统,这两种技术通常还是通过先后成像来实现关联的,但这种设计由于可确保方便地对样品中的同一个感兴趣区域进行成像,大幅降低了关联数据采集和分析的难度,为两者关联提供了便利条件.SRM-AFM关联最重要的作用是可以从样品表面和内部两方面提供特定目标的多重信息.例如, Odermatt等[94]将PALM-AFM关联应用于活细胞成像,先利用AFM监测活细胞前缘形貌随时间的变化(图13(b)),再利用PALM对同一部位进行超分辨成像,利用细胞中联接有光激活蛋白mEos2的桩蛋白实时成像结果(图13(c)—(g))在纳米尺度显示了细胞移动过程中其前缘黏着斑的动态变化.与SRM-EM关联类似,SRM-AFM关联也可用于对超分辨结果进行验证,例如Monserrate等[96]利用SMLM-AFM关联对荧光标记的离体λDNA进行成像,通过对比SMLM和AFM的成像结果,发现AFM图像中连续的结构在超分辨图像中存在间隙,从而反映超分辨样品标记和图像重构过程可能导致的伪影.此外,SRM-AFM关联还有另一种功能,即利用AFM对样品进行纳米操作并利用SRM技术进行成像甚至为纳米操作提供反馈信息[97].
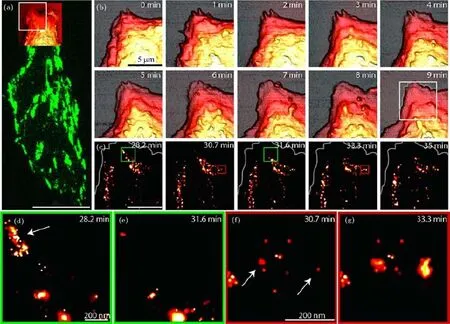
图13 SRM-AFM关联显微应用实例:提供内外结构信息[94]Fig.13.Correlative SRM-AFM applications:Providing both internal and external structural information[94].
4.3.3 超分辨-质谱关联显微
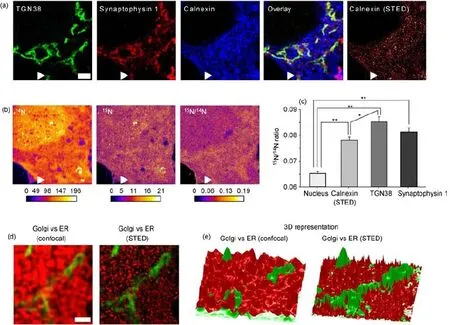
图14 SRM-SIMS关联显微应用实例:提供新的信息维度[99]Fig.14.Correlative SRM-SIMS applications:Providing additional information dimension[99].
质谱在同位素、原子团簇和分子的鉴别方面一直以来都扮演着非常重要的角色,而近几年利用二次离子质谱进行成像的二次离子质谱技术(secondary ion mass spectrometry,SIMS)则为生物样品的化学分析提供了一种新的方法,目前空间分辨率已达100 nm左右,与SRM技术基本相当[98].SIMS成像也需要在真空环境进行,因此SRM-SIMS关联面临着与SRM-EM关联同样的问题,即只有解决关联样品的制备问题才能实现有效的关联.例如,Saka等[99]利用STED-SIMS关联研究了体外培养海马神经元中的蛋白质周转,他们将含有同位素15N的标记物添加在神经元的培养液中,利用其代谢状态作为后期蛋白质周转速率的量化表征,接着他们对细胞进行固定、免疫荧光标记、脱水、树脂包埋、切片等处理,并对处理后的样品先后进行CLSM,STED和SIMS成像,通过分析CLSM和STED获得的细胞结构图像(图14(a))和SIMS获得的15N/14N密度比分布(图14(b))在超分辨细胞结构背景下对比了不同细胞器中蛋白质周转速率的不同(图14(c)).
5 总结和展望
光学显微成像由于系统孔径的衍射存在着分辨率极限,而近十几年来迅速发展的SRM技术采用各种巧妙的办法却可以“绕过”该限制获得远高于衍射极限分辨率的超分辨图像.其中,STORM, PALM等技术针对宽场成像系统进行空域处理,通过衍射极限范围内荧光分子先后发光和分时探测,实现单分子定位并重构超分辨图像;STED, RESOLFT等技术针对扫描成像系统进行空域处理,通过抑制激发斑中心点以外荧光分子发光,避免了激发斑尺寸对分辨率的影响而获得超分辨图像;SIM,SSIM等技术则从频域角度进行处理,利用摩尔条纹将样品的高频信息平移到系统OTF的截止频率范围内实现超分辨成像.
SRM技术将荧光显微成像的分辨率提高了近一个数量级,其取得的巨大发展为生物医学研究提供了强有力的工具;然而作为一种新兴技术,SRM技术在实际应用于生物医学研究时仍面临着许多亟待解决的问题,尤其是当研究对象是活的、具有一定厚度的、成分和结构复杂的细胞、组织等生物样品时.因此,除了进一步提高分辨率,如何实现SRM的多色成像、厚样品三维成像、活细胞快速成像等也是目前SRM领域的研究重点,这些问题的研究和解决将使SRM技术未来有可能为生物医学研究提供更有意义的活细胞实时超分辨图像和数据.
在SRM技术尚未十分完善的今天,将SRM与其他技术进行关联的超分辨关联显微是实现优势互补的最佳途径,成为近年来SRM的一个重要发展趋势.SRM技术与FM,CLSM,LTFM,FLIM等衍射受限光学显微技术的关联可弥补SRM目前的一些弱点,起到识别和区分特定目标、提供多层次背景结构信息、提高时间分辨能力、丰富功能成像信息、实现多色超分辨等作用,最大程度地发挥SRM在分辨率方面的优势;SRM与FS,SR-FTIR等光谱测量和成像技术的关联可大幅提高光谱成像技术的空间分辨率,同时为SRM增加额外的信息维度;SRM与EM,AFM,SIMS等非光学显微技术的关联,一方面能为同一个生物学问题提供不同尺度、不同维度或不同角度的信息互相印证,使研究更加深入,另一方面还能为验证SRM结果的正确性提供直接的证据.
然而,由于成像原理的不同,进行关联的两种显微成像技术对成像环境和样品制备的要求有可能大相径庭,尤其是SRM与EM等非光学显微技术的关联,尽管目前已经发展了许多关联样品制备方法和关联显微成像流程,但这一类关联显微的实际应用到目前为止仍然难度较大.同时,对于分辨率同样达到几十纳米甚至更高水平的两种或多种技术之间的关联,图像的对准和融合要求也非常高,因为不同方法获得的图像之间对准的精度必须控制在10 nm以内,否则由于没有精确对准而造成的伪影或分辨率降低等问题也会大幅削弱关联显微的意义.因此,只有深入地研究和解决这些问题,才能实现真正有效的超分辨关联显微并应用于生物医学研究.
需要补充的是,除了SRM技术与其他技术的关联,目前不同的SRM技术之间也在尝试进行关联显微,如SIM-SMLM等[100,101].而所有关联显微研究的目的都只有一个,即更好地发挥这些先进技术的优势,通过优势互补为生物医学领域提供更好的研究方法和手段.相信随着各种技术自身的进步,以及关联所涉及的样品制备、图像处理等相关技术的进一步发展完善,超分辨成像技术和超分辨关联显微技术将为生物医学研究提供越来越多的更有意义的数据,帮助人们更深入、更全面地理解生命和疾病,为科学技术和人类社会的发展提供帮助.
感谢深圳大学于斌教授关于衍射极限分辨率的讨论,感谢美国华盛顿大学化学系Joshua C.Vaughan教授和研究生Marco Howard,Lauren Gagnon关于各种超分辨成像技术的讨论.
[1]Volkmann H 1966 Appl.Opt.5 1720
[2]Airy G B 1835 Trans.Cambridge Philos.Soc.5 283
[3]Yu D Y,Tan H Y 2011 Engineering Optics(3rd Ed.) (Beijing:China Machine Press)p400(in Chinese)[郁道银,谈恒英 2011工程光学(第三版)(北京:机械工业出版社)第400页]
[4]Su X Y,Li J T,Cao Y P,Zhang Q C 2011 Information Optics(3rd Ed.)(Beijing:Science Press)p74(in Chinese)[苏显渝,李继陶,曹益平,张启灿2011信息光学(第三版)(北京:科学出版社)第74页]
[5]Betzig E,Patterson G H,Sougrat R,Lindwasser O W, Olenych S,Bonifacino J S,Davidson M W,Lippincott-Schwartz J,Hess H F 2006 Science 313 1642
[6]Rust M J,Bates M,Zhuang X 2006 Nat.Methods 3 793
[7]Hess S T,Girirajan T P K,Mason M D 2006 Biophys. J.91 4258
[8]Hell S W,Wichmann J 1994 Opt.Lett.19 780
[9]Klar T A,Jakops S,Dyba M,Hell S W 2000 Proc.Natl. Acad.Sci.USA 97 8206
[10]Gustafsson M G L 2000 J.Microsc.198 82
[11]Gustafsson M G L 2005 Proc.Natl.Acad.Sci.USA 102 13081
[12]Lin D,Ma W,Liu X,Sun Y,Wu Y 2008 Proc.2008 CISP 4 244
[13]Thompson R E,Larson D R,Webb W W 2002 Biophys. J.82 2775
[14]Betzig E 1995 Opt.Lett.20 237
[15]Dickson R M,Cubitt A B,Tsien R Y,Moerner W E 1997 Nature 388 355
[16]Bates M,Blosser T R,Zhuang X 2005 Phys.Rev.Lett. 94 108101
[17]Bates M,Huang B,Dempsey G T,Zhuang X 2007 Science 317 1749
[18]Heilemann M,van de Linde S,Schuttpelz M,Kasper R,Seefeldt B,Mukherjee A,Tinnefeld P,Sauer M 2008 Angew.Chem.Int.Ed.47 6172
[19]Huang B,Wang W,Bates M,Zhuang X 2008 Science 319 810
[20]Huang B,Jones S A,Brandenburg B,Zhuang X 2008 Nat.Methods 5 1047
[21]Pavani S R P,Thompson M A,Biteen J S,Lord S J, Liu N,Twieg R J,Piestun R,Moerner W E 2009 Proc. Natl.Acad.Sci.USA 106 2995
[22]Sharonov A,Hochstrasser R M 2006 Proc.Natl.Acad. Sci.USA 103 18911
[23]Giannone G,Hosy E,Levet F,Constals A,Schulze K, Sobolevsky A I,Rosconi M P,Gouaux E,Robert Tampé, Choquet D,Cognet L 2010 Biophys.J.99 1303
[24]Jungmann R,Steinhauer C,Scheible M,Kuzyk A,Tinnefeld P,Simmel F C 2010 Nano Lett.10 4756
[25]Jungmann R,Avendano M S,Woehrstein J B,Dai M, Shih W M,Yin P 2014 Nat.Methods 11 313
[26]Folling J,Bossi M,Bock H,Medda R,Wurm C A,Hein B,Jakobs S,Eggeling C,Hell S W 2008 Nat.Methods 5 943
[27]Dertinger T,Colyer R,Iyer G,Weiss S,Enderlein J 2009 Proc.Natl.Acad.Sci.USA 106 22287
[28]Ha fiN,Grunwald M,van den Heuvel L S,Aspelmeier T,Chen J H,Zagrebelsky M,Schütte O M,Steinem C, Korte M,Munk A,Walla P J 2014 Nat.Methods 11 579
[29]Chang H,Zhang M,Ji W,Chen J,Zhang Y,Liu B,Lu J,Zhang J,Xu P,Xu T 2012 Proc.Natl.Acad.Sci. USA 109 4455
[30]Quan T,Li P,Long F,Zeng S,Luo Q,Hedde P N, Nienhaus G U,Huang Z 2010 Opt.Exp.18 11867
[31]Pan D,Hu Z,Qiu F,Huang Z L,Ma Y,Wang Y,Qin L, Zhang Z,Zeng S,Zhang Y 2014 Nat.Commun.5 5573
[32]Chen C,Zong S,Wang Z,Lu J,Zhu D,Zhang Y,Cui Y 2016 ACS Appl.Mater.Interfaces 8 25825
[33]Zeng Z,Chen X,Wang H,Huang N,Shan C,Zhang H, Teng J,Xi P 2015 Sci.Rep.5 8359
[34]Chen X,Zeng Z,Wang H,Xi P 2015 Nano Res.8 2251
[35]Zhanghao K,Chen L,Yang X,Wang M,Jing Z,Han H, Zhang M Q,Jin D,Gao J,Xi P 2016 Light:Sci.Appl. 5 e16166
[36]Ni Y,Cao B,Ma T,Niu G,Huo Y,Huang J,Chen D, Liu Y,Yu B,Zhang M Q,Niu H 2017 eLife 6 e21660
[37]Lin D Y,Ma W Y 2007 Physics 36 783(in Chinese)[林丹樱,马万云2007物理36 783]
[38]Hell S W 1997 Topics in Fluorescence Spectroscopy: Nonlinear and Two-Photon-Induced Fluorescence(Vol. 5)(New York:Plenum)p361
[39]Wildanger D,Bückers J,Westphal V,Hell S W,Kastrup L 2009 Opt.Exp.17 16100
[40]Hell S W,Willig K I,Dyba M,Jakobs S,Kastrup L,Westphal V 2006 Handbook of Biological Confocal Microscopy(3rd Ed.)(New York:Pawley,Springer-Science+Business Media)p571
[41]Hell S W,Kroug M 1995 Appl.Phys.B:Lasers Opt.60 495
[42]Hofmann M,Eggeling C,Jakobs S,Hell S W 2005 Proc. Natl.Acad.Sci.USA 102 17565
[43]Bretschneider S,Eggeling C,Hell S W 2007 Phys.Rev. Lett.98 218103
[44]Donnert G,Keller J,Wurm C A,Rizzoli S O,Westphal V,Schönle A,Jahn R,Jakobs S,Eggeling C,Hell S W 2007 Biophys.J.92 L67
[45]Willig K I,Harke B,Medda R,Hell S W 2007 Nat. Methods 4 915
[46]Westphal V,Rizzoli S O,Lauterbach M A,Kamin D, Jahn R,Hell S W 2008 Science 320 246
[47]Berning S,Willig K I,Ste ff ens H,Dibaj P,Hell S W 2012 Science 335 551
[48]Liu Y,Ding Y,Alonas E,Zhao W,Santangelo P J,Jin D,Piper J A,Teng J,Ren Q,Xi P 2012 PLoS One 7 e40003
[49]Wang Y,Kuang C,Li S,Hao X,Xu Y,Liu X 2014 Microsc.Res.Techniq.77 935
[50]Yu J,Yuan J,Zhang X,Liu J,Fang X 2013 Chin.Sci. Bull.58 4045
[51]Yu W,Ji Z,Dong D,Yang X,Xiao Y,Gong Q,Xi P, Shi K 2016 Laser Photon.Rev.10 147
[52]Yan W,Yang Y,Tan Y,Chen X,Li Y,Qu J,Ye T 2017 Photon.Res.5 176
[53]Cai Z,Liu X,Jiang H,He D,Peng X,Huang S,Zhang Z 2015 Opt.Exp.23 25171
[54]Du Y,Huang J,Lin D,Niu H 2012 Anal.Bioanal.Chem. 404 793
[55]Heintzmann R,Jovin T M,Cremer C 2002 J.Opt.Soc. Am.A 19 1599
[56]Gustafsson M G L,Shao L,Carlton P M,Wang C J R, Golubovskaya I N,Cande W Z,Agard D A,Sedaty J W 2008 Biophys.J.94 4957
[57]Kner P,Chhun B B,Griffis E R,Winoto L,Gustafsson M G L 2009 Nat.Methods 6 339
[58]Shao L,Kner P,Rego E H,Gustafsson M G L 2011 Nat. Methods 8 1044
[59]Fiolka R,Shao L,Rego E H,Davidsonc M W,Gustafsson M G L 2012 Proc.Natl.Acad.Sci.USA 109 5311 [60]Planchon T A,Gao L,Milkie D E,Davidson M W,Galbraith J A,Galbraith C G,Betzig E 2011 Nat.Methods 8 417
[61]Chen B C,Legant W R,Wang K,Shao L,Milkie D E, Davidson M W,Janetopoulos C,Wu X S,Hammer J A,Liu Z,English B P,Mimori-Kiyosue Y,Romero D P,Ritter A T,Lippincott-Schwartz J,Fritz-Laylin L, Mullins R D,Mitchell D M,Bembenek J N,Reymann A,Böhme R,Grill S W,Wang J T,Seydoux G,Tulu U S,Kiehart D P,Betzig E 2014 Science 346 1257998
[62]Gao L,Shao L,Chen B,Betzig E 2014 Nat.Protoc.9 1083
[63]Li D,Shao L,Chen B,Zhang X,Zhang M,Moses B, Milkie D E,Beach J R,Hammer J A,Pasham M,Kirchhausen T,Baird M A,Davidson M W,Xu P,Betzig E 2015 Science 28 349
[64]Dan D,Lei M,Yao B,Wang W,Winterhalder M,Zumbusch A,Qi Y,Xia L,Yan S,Yang Y,Gao P,Ye T, Zhao W 2013 Sci.Rep.3 1116
[65]Jones S A,Shim S H,He J,Zhuang X 2011 Nat.Methods.8 499
[66]Yang Z,Sharma A,Qi J,Peng X,Lee D Y,Hu R,Lin D,Qu J,Kim J S 2016 Chem.Soc.Rev.45 4651
[67]Samuel D 1996 Science 273 488
[68]Caplan J,Niethammer M,Taylor R M,Czymmek K J 2011 Curr.Opin.Struct.Biol.21 686
[69]Watanabe S,Punge A,Hollopeter G,Willig K I,Hobson R J,Davis M W,Hell S W,Jorgensen Erik M 2011 Nat. Methods 8 80
[70]Xu K,Zhong G,Zhuang X 2013 Science 339 452
[71]Crossman D J,Hou Y,Jayasinghe I,Baddeley D,Soeller C 2015 Methods 88 98
[72]Barna L,Dudok B,Miczan V,Horvath A,Laszlo Z I, Katona I 2016 Nat.Protoc.11 163
[73]Huang F,Hartwich T M P,Rivera-Molina F E,Lin Y, Duim W C,Long J J,Uchil P D,Myers J R,Baird M A, Mothes W,Davidson,M W,Toomre Derek,Bewersdorf J 2013 Nat.Methods 10 653
[74]Lin D,Ma W,Duan S,Zhang Y,Du L 2006 Apoptosis 11 1289
[75]Bálint Š,Vilanova I V,Álvarez Á S,Lakadamyali M 2013 Proc.Natl.Acad.Sci.USA 110 3375
[76]Tam J,Cordier G A,Bálint Š,Álvarez Á S,Borbely J S,Lakadamyali M 2014 PLoS One 9 e115512
[77]Auksorius E,Boruah B R,Dunsby C,Lanigan P M P, Kennedy G,Neil M A A,French P M W 2008 Opt.Lett. 33 113
[78]Bückers J,Wildanger D,Vicidomini G,Kastrup L,Hell S W 2011 Opt.Exp.19 3130
[79]Wu Q,Qi J,Lin D,Yan W,Hu R,Peng X,Qu J 2017 Proc.SPIE 10069 1006922
[80]Zhang Z,Kenny S J,Hauser M,Li W,Xu K 2015 Nat. Methods 12 935
[81]Mlodzianoski M J,Curthoys N M,Gunewardene M S, Carter S,Hess S T 2016 PLoS One 11 e0147506
[82]Holman H Y N,Bechtel H A,Hao Z,Martin M C 2010 Anal.Chem.82 8757
[83]Whelan D R,Bell T D M 2015 ACS Chem.Biol.10 2874
[84]Giepmans B N G 2008 Histochem.Cell Biol.130 211
[85]van Rijnsoever C,Oorschot V,Klumperman J 2008 Nat. Methods 5 973
[86]de Boer P,Hoogenboom J P,Giepmans B N G 2015 Nat.Methods 12 503
[87]Hauser M,Wojcik M,Kim D,Mahmoudi M,Li W,Xu K 2017 Chem.Rev.117 7428
[88]Löschberger A,Franke C,Krohne G,van de Linde S, Sauer M 2014 J.Cell Sci.127 4351
[89]Wojcik M,Hauser M,Li W,Moon S,Xu K 2015 Nat. Commun.6 7384
[90]Betzig E,Trautman J K 1992 Science 257 189
[91]Sun Y,Lin D,Rui Y,Han D,Ma W 2006 J.Electron Microsc.55 165
[92]Ge G,Han D,Lin D,Chu W,Sun Y,Jiang L,Ma W, Wang C 2007 Ultramicroscopy 107 299
[93]Harke B,Chacko J V,Haschke H,Canale C,Diaspro A 2012 Opt.Nanoscopy 1 3
[94]Odermatt P D,Shivanandan A,Deschout H,Jankele R, Nievergelt A P,Feletti L,Davidson M W,Radenovic A, Fantner G E 2015 Nano Lett.15 4896
[95]Chacko J V,Zanacchi F C,Diaspro A 2013 Cytoskeleton 70 729
[96]Monserrate A,Casado S,Flors C 2014 ChemPhysChem 15 647
[97]Chacko J V,Canale C,Harke B,Diaspro A 2013 PLoS One 8 e66608
[98]Boxer S G,Kraft M L,Weber P K 2009 Annu.Rev. Biophys.38 53
[99]Saka S K,Vogts A,Krönert K,Hillion F,Rizzoli S O, Wessels J T 2014 Nat.Commun.5 3664
[100]Hamel V,Guichard P,Fournier M,Guiet R,Fluckiger I,Seitz A,Gonczy P 2014 Biomed.Opt.Exp.5 3326
[101]Rossberger S,Best G,Baddeley D,Heintzmann R,Birk U,Dithmar S,Cremer C 2013 J.Opt.15 094003
PACS:87.64.M—,42.40.Lx,42.30.Va,42.25.—p DOI:10.7498/aps.66.148703
Recent progress on super-resolution imaging and correlative super-resolution microscopy∗
Lin Dan-Ying Qu Jun-Le†
(Key Laboratory of Optoelectronic Devices and Systems of Ministry of Education and Guangdong Province,College of Optoelectronic Engineering,Shenzhen University,Shenzhen 518060,China)
19 April 2017;revised manuscript
21 June 2017)
The di ff raction of the fi nite aperture in the optical imaging system restricts further improvement of the resolution of optical microscopy,which is called the“di ff raction limit”.Since raised by Ernst Abbe in 1873,the problem of di ff raction limit has been one of the foci of academic research.In recent years,with the rapid development of related f i elds such as the development of optoelectronic devices including high energy lasers and high sensitivity detectors and the development of new fl uorescent probes,the problem of di ff raction limit in optical microscopy ushered in a new opportunity,and super-resolution microscopy(SRM)has made remarkable achievements in the past decade.The basic principles of di ff raction limited resolution in both space and frequency domains are reviewed,and on this basis, the mechanisms for the various SRM technologies to circumvent the di ff raction limit and improve the resolution are explained in detail.The development trends and research directions of various SRM techniques are also introduced. As a new and important development trend of SRM,correlative super-resolution microscopy and its recent progress are reviewed,including correlative studies on SRM and time-lapse live cell fl uorescence microscopy, fl uorescence lifetime imaging microscopy,spectrometry and spectroscopy,electron microscopy,atomic force microscopy,etc.The role and signi fi cance of various correlative super-resolution microscopy are discussed.The future development of super-resolution microscopy and correlative super-resolution microscopy is also prospected.
di ff raction limit,super-resolution imaging,correlative microscopy,correlative superresolution microscopy
:87.64.M—,42.40.Lx,42.30.Va,42.25.—p
10.7498/aps.66.148703
∗国家重点基础研究发展计划(批准号:2015CB352005)、国家自然科学基金(批准号:61525503,61378091,61620106016)和广东省自然科学基金创新研究团队项目(批准号:2014A030312008)资助的课题.
†通信作者.E-mail:jlqu@szu.edu.cn
©2017中国物理学会Chinese Physical Society
http://wulixb.iphy.ac.cn
*Project supported by the National Basic Research Program of China(Grant No.2015CB352005),the National Natural Science Foundation of China(Grant Nos.61525503,61378091,61620106016),and the Natural Science Foundation of Guangdong Province for Innovation Research Team,China(Grant No.2014A030312008).
†Corresponding author.E-mail:jlqu@szu.edu.cn
猜你喜欢
新世纪智能(数学备考)(2021年9期)2021-11-24
装备制造技术(2020年1期)2020-12-25
当代陕西(2019年15期)2019-09-02
数学物理学报(2019年3期)2019-07-23
家庭影院技术(2018年9期)2018-11-02
学苑创造·A版(2018年11期)2018-02-01
制造技术与机床(2017年7期)2018-01-19
自动化学报(2017年5期)2017-05-14
读者(2017年5期)2017-02-15
现代检验医学杂志(2016年3期)2016-11-15
