
晚发型脊柱骨骺发育不良伴进行性骨关节病1家系报道及基因研究
2017-08-07 06:56刘书中李政垚李子全王牧川王以朋
中国实验诊断学 2017年7期
刘书中,宋 桉,李政垚,李子全,王牧川,王以朋*
(中国医学科学院 北京协和医学院 北京协和医院 1.骨科;2.内分泌科,北京100730)
*通讯作者
晚发型脊柱骨骺发育不良伴进行性骨关节病1家系报道及基因研究
刘书中1,宋 桉2,李政垚1,李子全1,王牧川1,王以朋1*
(中国医学科学院 北京协和医学院 北京协和医院 1.骨科;2.内分泌科,北京100730)
晚发型脊柱骨骺发育不良(spondyloepiphyseal dysplasia,SED)系一组同时累及脊柱和长管状骨骨骺的遗传性发育障碍性疾病,临床上有躯干与肢体不成比例的矮小身材特征。晚发型SED发病率低,约为0.1-0.4/10万,是一种较为罕见的骨发育不良疾病[1]。
1 临床资料
患者,女,31岁,因间断性全身多关节疼痛25年、加重5年就诊。该患者6岁前发育正常,6岁后无明显诱因出现双侧髋部疼痛,程度逐渐加重,并逐渐出现双膝、双肘、双侧指间关节、双腕、双肩疼痛及活动受限,发育较同龄人迟缓,病
程中无晨僵,无腰背痛、足跟痛,无腹痛、腹泻、尿频、尿急、眼部不适等表现。查体:身高145 cm,智力正常,短颈,肩峰高耸,桶状胸。脊柱前屈、后伸、左右侧屈均明显受限,双髋关节内旋受限,双侧肩、肘、腕关节活动明显受限,双肘关节屈曲畸形,爪形手,双侧骶髂关节压痛(+),双侧4字试验(+)。实验室检查:血尿便常规、红细胞沉降率、C反应蛋白、肝肾功、电解质、凝血功能均无显著异常,免疫球蛋白、补体、RF、ASO、HLA-B27、ACL、ANA、抗CCP抗体均(-)。脊柱正侧位X线:椎体横径增大,两侧缘低平,中央部膨突,见图1。四肢长骨X线检查:长管状骨明显短缩,干骺端扩张,结构紊乱,不均匀致密,边缘呈刺状突出。骨骺形态不规则,边缘不规整,部分呈碎裂状,胫骨两侧向内倾斜,呈“O”形改变。双手正位:双侧指间关节间隙狭窄,骨性膨大,屈曲畸形。骨盆正位:双髋关节间隙狭窄,股骨头塌陷变形,见图2。双膝X线:双膝关节间隙狭窄,骨质增生。家系调查:家族中无近亲结婚,母亲孕期无患病或服药史。进一步对该患者家系进行了详细调查,绘制了家系图谱(见图3),该家系3代中2例患者均为女性,先证者为该患者姐姐,发病年龄均在6-8岁之间。
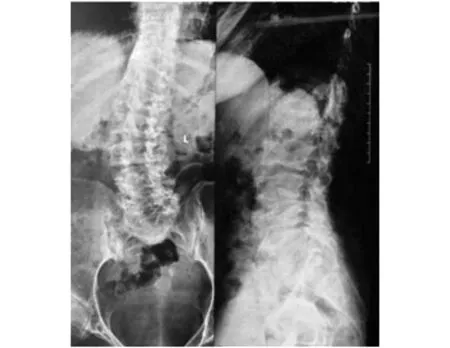
图1 先证者脊柱正侧位X线片

图2 先证者骨盆正位X线片
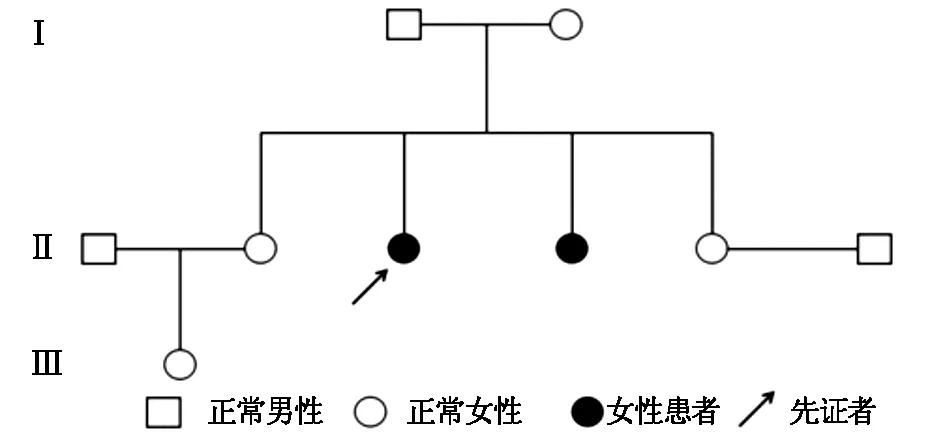
图3 患者家系遗传图谱
2 对象和方法
经患者及家属知情同意后,采集家系成员外周血6份(10 ml/人),同时抽取30名无关志愿者静脉血液进行对照试验,其中男10名,女20名。之后采用TIANamp Blood DNA Kit提取基因组DNA,记录DNA浓度后,EP管密封-4℃保存备用。设计引物扩增WISP3等363个基因。扩增步骤为:预变性温度为94℃ 3分钟,94℃ 55秒,64℃ 55秒,72℃ 60秒,35个循环,最后延伸为72℃ 10分钟,PCR产物用1.5%琼脂糖凝胶电泳分离,溴乙锭染色后观察条带。使用相同的引物,测序仪器为ABI PRISM 3730,测序试剂为BigDye terminator v3.1,由天津华大基因生物公司测序部对PCR扩增产物进行纯化及测序。将突变片段的测序结果与GeneBank中363个基因外显子DNA序列进行比较。
3 结果
基因检测检出该家系中两位患者均携带WISP3基因的2个杂合突变c.396T>G(p.Cys132Trp)和c.721T>G(p.Cys241Gly),c.396T>G(p.Cys132Trp)和c.721T>G(p.Cys241Gly)均未经文献报道,可分别导致WISP3基因编码的第132位半胱氨酸突变为色氨酸以及第241位半胱氨酸突变为甘氨酸。在家系中正常个体及30名对照无关志愿者中未发现上述两个基因突变存在。
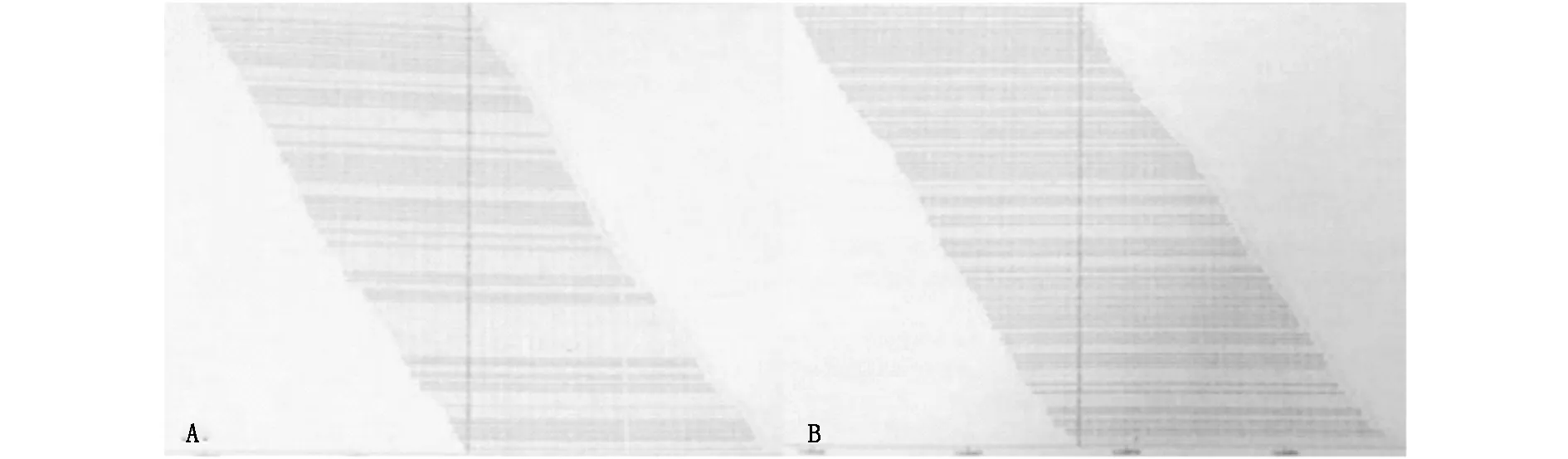
图4 (A)WISP3基因突变c.396T>G(p.Cys132Trp)支持图。(B)WISP3基因突变c.721T>G(p.Cys241Gly)支持图
4 讨论
SED根据发病时间不同可分为早发型(先天型)和晚发型(迟发型)两种[2]。早发型SED系常染色体显性遗传病,患儿出生后即见异常。晚发型SED患者通常在5岁以后出现症状,其遗传方式包括X染色体隐性遗传(仅男性发病)或常染色体显性及隐性遗传(男女均可发病),本型在临床上主要以短躯干侏儒、腰部与四肢大关节疼痛及活动受限为特征。本型主要X线特征包括扁平椎、椎间隙狭窄或增宽、脊柱前侧凸、髋内翻畸形、股骨远端及肱骨近端干骺端变宽而不规则并可伴有骨赘形成等[3]。随病变进展,全身多关节疼痛及活动受限逐渐加重,本家系中2例患者临床特点均与之相符。
临床上晚发型SED需与以下疾病进行鉴别:①黏多糖病IV型:是常染色体隐性遗传疾病,发病常与近亲结婚有关,此外尚有肝脾肿大、角膜混浊、耳聋、尿黏多糖呈强阳性等特异表现,X线中“子弹头样椎体”和“飘带样肋骨”是其特征性影像学改变,尿筛查试验及血酶活性测定可予鉴别[4]。②Scheuermann病(椎体骺板缺血性坏死):常在10岁左右发病,可侵犯全部胸腰椎,椎体前缘楔形变窄,典型者呈阶梯状,但无骨盆及椎间隙狭窄改变[5]。此外,还需要进行鉴别的疾病包括成骨不全综合征、脊柱干骺端发育不良、多发性骨骺发育不良及幼年型类风湿关节炎等。
诊断方面,晚发型SED诊断的确立需结合患者发病特征、临床表现、特征影像学改变以及基因测序,在疾病问诊中需特别注意对家族史的询问。该家系中两位患者均携带WISP3基因的2个杂合突变c.396T>G(p.Cys132Trp)和c.721T>G(p.Cys241Gly),二者均未经文献报道,可分别导致WISP3基因编码的第132位半胱氨酸突变为色氨酸以及第241位半胱氨酸突变为甘氨酸,而在家系中正常个体及30名对照无关志愿者中未发现上述两个基因突变存在,WISP3相关的晚发型脊柱骨骺发育不良伴进行性骨关节病为常染色体隐性遗传,纯合或复合杂合基因突变可导致疾病发生,因此,c.396T>G(p.Cys132Trp)和c.721T>G(p.Cys241Gly)突变很可能形成复合杂合形式,进而与致病相关[6-8]。
治疗方面,目前针对晚发型SED尚缺乏有效的治疗手段。对于晚发型SED患者,在疾病早期可予适当抗骨质疏松、营养软骨、消炎镇痛治疗,但长期疗效有待进一步论证。对于晚期SED患者出现全身严重畸形及功能障碍时,为提高患者生活质量、改善畸形,可酌情行假体置换或人工关节置换术[9]。近年来,晚发型SED相关致病基因位点不断被发现,使得在分子水平对晚发型SED进行诊断及治疗成为可能,但尚需进一步深入研究晚发型SED的遗传特点,以期将新的诊疗技术早日应用于临床造福该病患者。
[1]GedeonAK,ColleyA,Jamieson R,et al.Identification of the gene(SEDL) causing X-linked spondyloepiphysealdysplasia tarda[J].NatGenet,1999,22(4):400.
[2]Kocyigit H,Arkun R,Ozkinay F,et al.Spondyloepiphyseal dysplasia tarda with progressive arthropathy[J].Clin Rheumatol,?2000,19(3):238.
[3]张 华,杜联军,张 欢,等.脊柱骨骺发育不良的影像学表现[J].诊断学理论与实践,2004,3(3):182.
[4]杨 波,金 今,翁习生,等.6例脊柱骨骺发育不良患者的诊断及治疗[J].中国骨与关节外科,2008,1(4):285.
[5]孙 静,夏维波,李 梅,等.迟发性脊柱骨骺发育不良的临床诊断及SEDL基因突变分析[J].中华骨质疏松和骨矿盐疾病杂志,2012,5(1):7.
[6]Liu L,Li N,Zhao Z,et al.Novel WISP3 mutations causing spondyloepiphyseal dysplasiatardawithprogressive arthropathy in two unrelated Chinese families[J].Joint Bone Spine,2015,82(2):125.
[7]Jurgens J,Sobreira N,Modaff P,et al.Novel COL2A1 variant (c.619G>A,p.Gly207Arg) manifestingas a phenotype similar to progressivepseudorheumatoid dysplasiaandspondyloepiphysealdysplasia,Stanescu type[J].Hum Mutat,2015,36(10):1004.
[8]Montané LS,Marín OR,Rivera-Pedroza CI,et al.Early severe scoliosis in a patient with atypicalprogressive pseudorheumatoid dysplasia(PPD):Identification of two WISP3 mutations,one previously unreported[J].Am J Med Genet A,2016,170(6):1595.
[9]Kim RH,Scuderi GR,Dennis DA,et al.Technical challenges of total knee arthroplasty inskeletaldysplasia[J].Clin Orthop Relat Res,2011,469(1):69.
1007-4287(2017)07-1225-03
2016-07-24)
猜你喜欢
中华实用诊断与治疗杂志(2022年1期)2022-08-31
中华实用诊断与治疗杂志(2022年1期)2022-08-31
昆明医科大学学报(2021年1期)2021-02-07
中国生殖健康(2020年2期)2021-01-18
中华养生保健(2020年5期)2020-11-16
中华养生保健(2020年4期)2020-11-16
中南林业科技大学学报(2019年4期)2019-04-08
小学生导刊(2018年13期)2018-06-29
森林工程(2018年1期)2018-05-14
中国生殖健康(2018年2期)2018-01-12
