
液相色谱-串联质谱法测定水产品中磺胺类及喹诺酮类药物残留量
2017-07-20 10:09李婵君李柱梅秦喜玲董素静王彦超
理化检验-化学分册 2017年5期
李婵君,李柱梅,秦喜玲,董素静,王彦超
(郑州市农产品质量检测流通中心,郑州450006)
液相色谱-串联质谱法测定水产品中磺胺类及喹诺酮类药物残留量
李婵君,李柱梅,秦喜玲,董素静,王彦超
(郑州市农产品质量检测流通中心,郑州450006)
水产品样品(5.00 g)经乙腈-甲酸(99+1)混合液20 mL提取,无水乙醇除水,浓缩并加正己烷2 mL脱脂。所得溶液进行液相色谱分离。以ACQUITY UPLC BEH HILIC色谱柱为分离柱,以不同体积比的甲醇和0.1%(体积分数)甲酸溶液的混合液为流动相进行梯度洗脱。质谱分析中,采用电喷雾正离子源多反应监测模式检测。采用内标法定量。所涉11种药物的线性范围均为5~200μg·L-1,方法的测定下限(10S/N)在0.07~0.20μg·kg-1之间。在1.0,4.0,20.0μg·kg-1等3个浓度水平进行加标回收试验,回收率在80.3%~119%之间,测定值的相对标准偏差(n=6)在1.3%~12%之间。
液相色谱-串联质谱法;磺胺类;喹诺酮类;水产品
喹诺酮类和磺胺类药物为常见合成抗菌剂,在畜禽和水产养殖中使用十分普遍,但是由于存在着药物滥用、误用及不遵守休药期规定等现象,造成很多动物源性食品中残存各种药物及药物的代谢物,从而对人的身体健康构成威胁或直接造成损害。测定此类药物的方法目前有酶联免疫法[1]、薄层色谱法[2]、液相色谱法[3-8]和液相色谱-质谱法[9-12]等。
本工作在农业部1077号公告-1-2008的基础上,优化了试验条件,建立了液相色谱-串联质谱法测定水产品中磺胺类和喹诺酮类11种抗生素含量的方法。以2种氘代磺胺试剂(氘代磺胺邻二甲氧嘧啶、氘代磺胺间二甲氧嘧啶)和3种氘代喹诺酮试剂(氘代诺氟沙星、氘代恩诺沙星、氘代环丙沙星)作内标,采用标准加入法进行定量分析,为水产品中药物残留检测提供了新思路。
1 试验部分
1.1 仪器与试剂
Waters Xevo TQ-S型超高压液相色谱-串联质谱仪;SIGMA 3K15型高速冷冻离心机;BUCHI R-210型旋转蒸发仪;Milli-Q Advantage A10型纯水机。
诺氟沙星、恩诺沙星、环丙沙星标准储备溶液: 1.000 g·L-1,分别称取诺氟沙星、恩诺沙星、环丙沙星(纯度不小于98%)10 mg于不同的10 mL容量瓶中,加甲酸0.2 mL,用甲醇溶解并稀释至刻度。避光-18℃下保存,逐级稀释备用。
磺胺二甲氧嘧啶、磺胺二甲基嘧啶、磺胺二甲异噁唑、磺胺甲基嘧啶、磺胺噻唑、磺胺甲噁唑、磺胺嘧啶标准储备溶液:1.000 g·L-1,分别称取磺胺二甲氧嘧啶、磺胺二甲基嘧啶、磺胺二甲异噁唑、磺胺甲基嘧啶、磺胺噻唑、磺胺甲噁唑、磺胺嘧啶(纯度不小于98%)10 mg于不同的10 mL容量瓶中,用甲醇溶解并稀释至刻度。避光-18℃下保存,逐级稀释备用。
磺胺喹噁啉标准储备溶液:1.000 g·L-1,称取磺胺喹噁啉(纯度不小于98%)10 mg于10 mL容量瓶中,加入氨水20μL,用甲醇溶解并稀释至刻度。避光-18℃下保存,逐级稀释备用。
氘代诺氟沙星、氘代恩诺沙星、氘代环丙沙星内标储备溶液:0.500 g·L-1,分别称取氘代诺氟沙星、氘代恩诺沙星、氘代环丙沙星(纯度不小于98%)5 mg于不同的10 mL容量瓶中,加甲酸0.2 mL,用甲醇溶解并稀释至刻度。避光-18℃下保存,逐级稀释备用。
氘代磺胺邻二甲氧嘧啶、氘代磺胺间二甲氧嘧啶内标储备溶液:0.500 g·L-1,分别称取氘代磺胺邻二甲氧嘧啶、氘代磺胺间二甲氧嘧啶(纯度不小于98%)5 mg于不同的10 mL容量瓶中,用甲醇溶解并稀释至刻度。避光-18℃下保存,逐级稀释备用。
混合内标溶液:1 mg·L-1,将0.5 g·L-1各内标储备溶液混合稀释配制而成。
乙腈、正己烷、甲酸为色谱纯;试验用水为一级纯水。
1.2 仪器工作条件
1)色谱条件ACQUITY UPLC BEH HILIC色谱柱(2.1 mm×100 mm,1.7μm);柱温40℃;流量0.3 mL·min-1;进样量5μL。流动相:A为甲醇,B为0.1%(体积分数,下同)甲酸溶液。梯度洗脱程序:0~0.5 min时,A为5%;0.5~5 min时,A由5%升至95%;5.01 min时,A由95%跳转至5%,保持2 min。
2)质谱条件电喷雾离子源,正离子扫描,离子源温度150℃;多反应监测(MRM)模式;毛细管电压1.0 kV;脱溶剂温度500℃,脱溶剂气流量1 000 L·h-1。其余质谱参数见表1,其中“*”为定量离子。

表1 质谱参数Tab.1 MS parameters

表1 (续)
1.3 试验方法
称取试样5.00 g于50 mL离心管中,加入1 mg·L-1混合内标溶液50μL,漩涡混合30 s,避光放置10 min。再加入乙腈-甲酸(99+1)混合液20 mL,涡流混合1 min后再超声波提取10 min。以4 000 r·min-1转速离心5 min,取上清液于50 mL梨形瓶中,残渣中加入乙腈-甲酸(99+1)混合液20 mL,重复提取一次,合并两次提取液,于40℃水浴旋转蒸发至2~3 mL,加入无水乙醇10 mL,旋转蒸发至近干。加20%(体积分数)甲醇溶液1.0 mL漩涡超声溶解残留物,再加入正己烷2.0 mL漩涡混合30 s,转入5 mL具塞离心管中,然后以4 000 r· min-1转速离心5 min,弃上层液,取下清液,过0.2 μm滤膜,按仪器工作条件进行测定。
2 结果与讨论
2.1 前处理条件的选择
磺胺类化合物在酸性条件下溶解度较大,使用酸化乙腈提取,可提高提取效率。
样品中含有水分,在样品提取液旋转蒸发时,会出现很难蒸干的情况,加入无水硫酸钠可除去样品中水分,但影响喹诺酮类药物提取率;可在旋蒸至2~3 mL时,加入无水乙醇10 mL,使样品提取液蒸至近干,且喹诺酮类提取效率稳定,多次试验重复性较好。
2.2 质谱条件的选择
在正离子模式下,11种化合物及5种内标均可获得较高的[M+H]+母离子,通过调节仪器的毛管电压、锥孔电压等参数,使目标物质的响应信号最高。流动相中A分别为乙腈、甲醇,B分别为含0.1%甲酸的5 mmol·L-1乙酸铵溶液、含0.1%甲酸的2 mmol·L-1乙酸铵溶液、0.1%甲酸溶液,以不同比例的A、B作为流动相。结果发现:A为甲醇时,可提高目标物灵敏度,且峰形更好;B为0.1%甲酸溶液时,得到同样离子化效果。试验选用甲醇和0.1%甲酸溶液作为流动相。各化合物离子流图见图1。
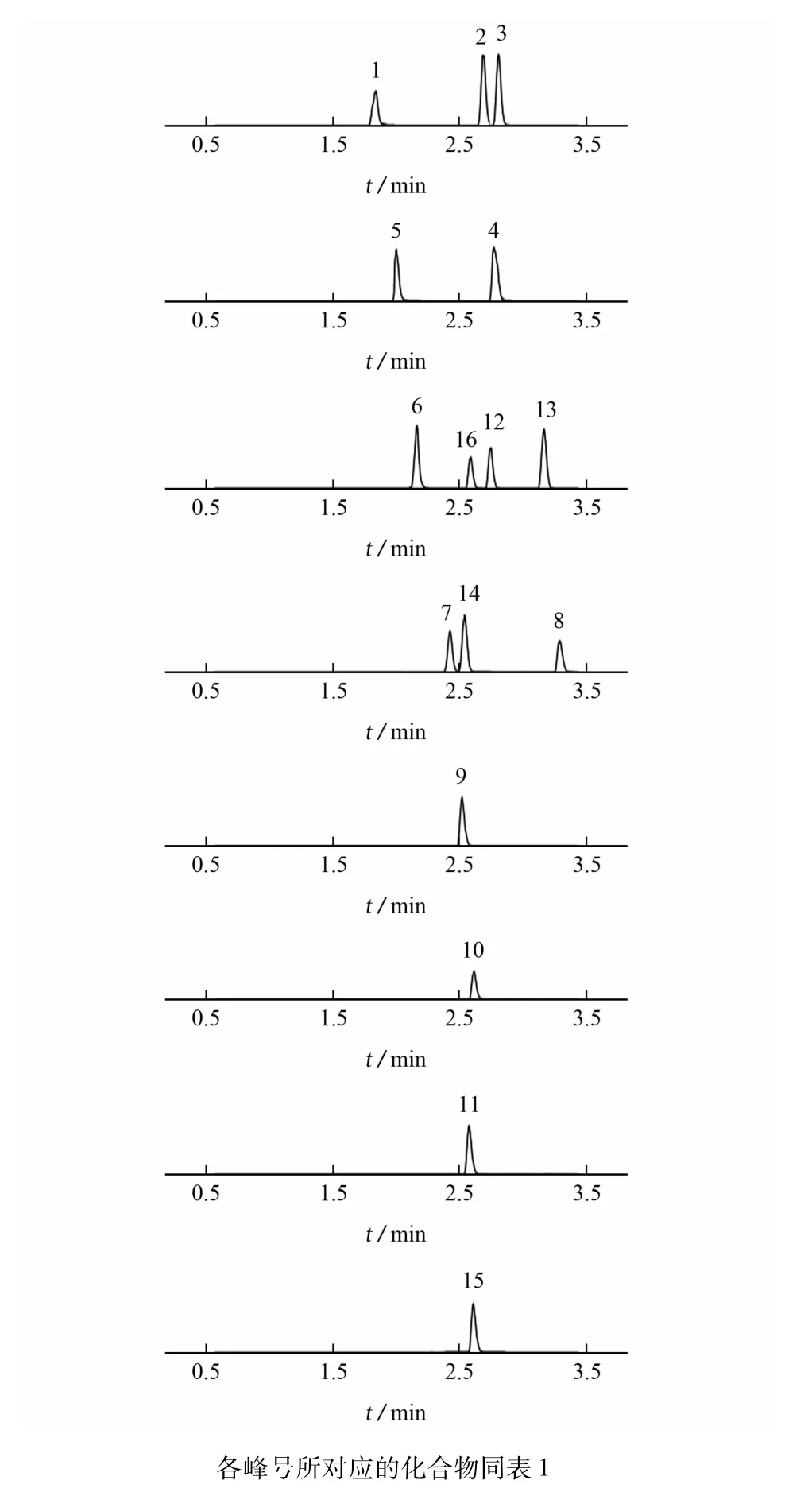
图1 选择离子流色谱图Fig.1 SIC chromatograms
2.3 基质效应
水产品基质复杂,用液相色谱-质谱法测定基质效应显著,特别是磺胺类药物基质抑制效应较强,减少基质效应影响的方法一般有加入同位素标记的目标物、基质配制标准溶液、更多的净化步骤或进样前稀释等[8]。
试验采用内标法,在此基础上用试剂配制标准溶液(方法1)和基质配制标准溶液(方法2),向空白样品加入标准品后按试验方法进行测定(方法3,简称标准加入法);进行加标试验,低添加,采用3种方法绘制标准曲线,所添加量在线性范围内,每个添加进行3次平行试验,6批次重复性试验。用方法1定量:添加质量分数为4.0μg·kg-1时,无法定量;质量分数为10.0μg·kg-1时,沙星类均可定量且回收率为91.0%~115%,磺胺类回收率为20%~55%,且重复性不好。采用方法2定量:添加质量分数为1.0μg·kg-1时,回收率在44%~62%之间,且重复性不好;采用加入内标物,标准加入法定量时,可以准确定量微量添加,添加质量分数为0.20μg·kg-1,最终计算实际添加质量分数在0.162~0.234μg·kg-1内,回收率在81.5%~118%之间,相对标准偏差(RSD)为3.4%~12%。试验选用标准加入法。
采用标准加入法内标法定量能在线性范围内准确定量,适合微量添加,测定结果稳定。在样品前处理过程中,待测物会有不同程度的损失,特别是对于含量较低的样品,这种损失特别明显,因此采用前两种方法定量时,含量较低的样品,不能定量或定量不准确;采用标准加入法加入内标物,和待测样品经过同样的前处理过程,可进行样品微量分析且定量更准确,标准曲线的线性范围更具有实用性。
2.4 标准曲线与测定下限
称取空白样品6份各5.00 g,分别加入不同体积的1 mg·L-111种化合物的标准溶液及1 mg· L-15种内标溶液50μL,配制成质量浓度分别为5,10,20,50,100,200μg·L-1的混合标准溶液。
磺胺二甲氧嘧啶、磺胺喹噁啉、磺胺二甲异噁唑以氘代磺胺间二甲氧嘧啶为内标;磺胺二甲基嘧啶、磺胺甲基嘧啶、磺胺噻唑、磺胺甲噁唑、磺胺嘧啶以氘代磺胺邻二甲氧嘧啶为内标;诺氟沙星以氘代诺氟沙星为内标;恩诺沙星以氘代恩诺沙星为内标;环丙沙星以氘代环丙沙星内标;用内标法绘制标准曲线,结果表明:11种化合物的线性范围为5~200μg·L-1,线性回归方程、相关系数见表2。
按10倍信噪比计算各化合物的测定下限(10S/N),其结果见表2。

表2 线性回归方程、相关系数和测定下限Tab.2 Linear regression equations,correlation coefficients and lower limits of determination
2.5 样品分析
抽取市售鲤鱼、草鱼、鲫鱼、黄鱼、鳜鱼各4条,共20个样品,按试验方法进行测定,未检出目标物。在样品中加入11种化合物的混合标准溶液,添加水平为1.0,4.0,20.0μg·kg-1,每个加标样品平行测定6次,精密度和回收试验结果见表3。

表3 精密度和回收试验结果(n=6)Tab.3 Results of tests for precision and recovery(n=6)

表3 (续)
本工作采用液相色谱-串联质谱法同时测定磺胺类和喹诺酮类共11种抗生素的含量,以标准加入法进行定量分析,减小基质干扰,提高了检测准确度,方法具有较高的灵敏度、精密度和较低的定量限,线性范围宽,在实际样品检测中分析速率快,稳定性好,结果准确可靠。
[1]HOHZAPPLE C K,BUCKLEY S A,STANKER L H.Production and characterization of monoclonal antibodies againstsarafioxacin and cross-reactivity studies of related fiuoroquinolones[J].J Chromatogr B,2001,754(1):1-9.
[2]JUHEL-GAUGAIN M,ABJEAN J P.Screening of quinolone resvidues in pig muscle by planar chromatography[J].Chrornatographia,1998,47:101-104.
[3]ZENG Z,DONG A,RANG G,et al.Simultaneous determination of nine fluoroquinolones in egg white and egg yolk by liquid chromatography with fluorescence detection[J].J Chromatogr B,2005,821(2):202-209.
[4]杜黎明,卫洪清,张俊燕,等.反相高效液相色谱法同时测定6种氟喹诺酮类药物[J].色谱,2003,5(7): 503-506.
[5]胡献刚,罗义,周启星,等.固相萃取-高效液相色谱法测定畜牧粪便中13种抗生素药物残留[J].分析化学,2008,36(9):1162-1166.
[6]刘亚梅,闵盛.高效液相色谱法同时测定禽蛋中环丙沙星、达氟沙星和恩诺沙星的残留量[J].理化检验-化学分册,2016,52(3):305-307.
[7]孟勇,张美琴,王静,等.高效液相色谱法测定水产品中7种兽药残留量[J].理化检验-化学分册,2012,48 (5):543-546.
[8]张林田,黄少玉,陈小雪,等.高效液相色谱法测定水产品中四种氟喹诺酮类药物残留[J].理化检验-化学分册,2009,45(9):1086-1087.
[9]TOUSSAINT B,BORDIN G,JANOSI A,et al.Validation of a liquid chromatography-tandem mass spectrometry method for the smiultaneous quantification of11(fluoro) quinolone antibiotics in swine kidney[J].J Chromatogr A,2002,976(1/2):195-206.
[10]LIM J,PARK B,YUN H.Sensitive liquid chromautographic.mass spectrometry assay for norfloxacin in poultry tissue[J].J Chromatogr B,2002,772:185-189.
[11]王立琦,贺利民,曾振灵,等.液相色谱-串联质谱检测兽药残留中的基质效应的研究进展[J].质谱学报,2011,32(6):321-332.
[12]谢寒冰,蒋万枫,赵海峰,等.高效液相色谱-四极杆飞行时间质谱法测定猪肉中20种兽药残留量[J].理化检验-化学分册,2014,50(6):702-707.
LC-MS/MS Determination of Residual Amounts of Sulfonamides and Quinolones in Aquatic Products
LI Chanjun,LI Zhumei,QIN Xiling,DONG Sujing,WANG Yanchao
(Testing Center for Quality of Agricultural Produsts of Zhengzhou,Zhengzhou 450006,China)
The sample of aquatic products(5.00 g)was extracted with 20 mL of a mixture of acetonitrile and formic acid(99+1).The extract was dehydrated with absolute ethanol.The extract was then concentrated and 2 mL of nhexane was added to remove fat.LC separation was performed by using ACQUITY UPLC BEH HILIC chromatographic column as stationary phase,and mixtures of methanol and 0.1%(φ)formic acid solution with various ratios as mobile phase in gradient elution.ESI+and multi-reaction-monitoring were adopted in MS/MS.Internal standard method was used for quantification.Linearity ranges of the 11 drugs were found in the same range of5-200μg·L-1,with lower limits of determination(10S/N)in the range of0.07-0.20μg·kg-1.Tests for recovery were made by standard addition method at3 concentration levels of1.0,4.0,20.0μg·kg-1,giving values of recovery and RSD's(n=6)in the ranges of80.3%-119%and 1.3%-12%respectively.
LC-MS/MS;sulfonamides;quinolones;aquatic products
O657.63
A
1001-4020(2017)05-0527-05
10.11973/lhjy-hx201705007
2016-05-11
李婵君(1978-),女,河南新密人,主要从事农产品质量检测工作,chanjunli@163.com
猜你喜欢
昆明医科大学学报(2021年8期)2021-08-13
昆明医科大学学报(2021年3期)2021-07-22
农药科学与管理(2019年6期)2019-11-23
武警医学(2018年10期)2018-11-06
环境保护与循环经济(2017年10期)2017-03-16
国外医药(抗生素分册)(2016年5期)2016-07-12
国外医药(抗生素分册)(2016年2期)2016-07-12
湖南农业(2016年12期)2016-03-10
现代农业(2016年4期)2016-02-28
中国药物经济学(2013年1期)2013-05-05
