
基于静电势电荷的抗击埃博拉病毒新药物法匹拉韦及其衍生物水溶解度logS值预测
2017-07-10 22:43裴诗恩黄颖琦吴淑曼彭自珍苏凌峰刘喜灵陈晗剑钟爱国
当代化工 2017年1期
裴诗恩 黄颖琦 吴淑曼 彭自珍 苏凌峰 刘喜灵 陈晗剑 钟爱国


摘要:运用分子中的原子理论(AIM),探讨了以法匹拉韦分子羰基氧原子值为目标,测试了不同基组和泛函选择的依赖性。然后用密度泛函理论(DFT B3LYP)和6-31+G(d,p)基组,优化了20种法匹拉韦及其常见衍生物的分子结构,分别得到11号羰基氧的密立根电荷(MUL-O)、自然原子轨道电荷(NBO-O)、何秀巴赫电荷(HIR-O)和静电势电荷(EsP-O)值,发现11号氧原子的ESP-O电荷值与用ACD Lab6.0预测出来的logS值相关性最好,相关系数达0.986;计算了法匹拉韦及其11种未知衍生物的ESP-O电荷值,代入相关最佳线性方程,发现所得结果与ACD Lab6.0预测结果十分接近,最大误差绝对对数值仅为0.08;分子的静电势图也显示法匹拉韦及其甲基法匹拉韦发挥其药理毒理作用可能的部位在电负性强的羰基氧原子上。
关键词:密度泛函理论;法匹拉韋;Mulliken电荷;NBO电荷;ESP电荷;logS值
中图分类号:O 644.32 文献标识码:A 文章编号:1671-0460(2017)01-0031-04
法匹拉韦(Favipiravir,商品名为ANIGAN,T705,),化学名为6-氟-3-羟基吡嗪-2-甲酰胺,是日本富山化学有限公司研制的新型RNA依赖的RNA聚合酶抑制类广谱抗病毒药,2011年3月在日本完成Ⅲ期临床试验,临床可用于治疗流感。法匹拉韦和其他抗击流感病毒药物的作用机理不同,它主要通过阻断病毒核酸复制的方法来抑制病毒繁殖。在2014年抗击埃博拉病毒疫情的战役中,日本Fujifilm公司最近获批的流感治疗药物法匹拉韦可以对抗西非埃博拉病毒的漫延,法匹拉韦作为明星药物也确实在治疗埃博拉患者中取得良好的效果。分子酸碱度pKb值和溶解度值logS是药物分子的一个基本物化性质,其对人体的药物吸收程度有重要影响。通常它的值可以通过实验来测定,但实验测定方法会有一定的局限性,在实际使用中,由于分子受稳定性等诸多因素的制约,使从实验上很难准确测定一些分子的pKb值和溶解度值,尤其是像法匹拉韦这些最新出现的药物。因此从理论和计算上寻找有效和可靠的预测酸碱电离性的方法成为目前十分活跃的课题。
当前已有多种方法用于预报有机化合物的脂水分配系数logP值和水溶解度logS值。相比较而言,脂水分配系数logP值的计算方法更为成熟和有效些。这是因为,相对于溶质在水和正辛醇两种液-液之相间的分配,分子溶质的溶解过程则是更为复杂的物化过程,它不仅有溶质一溶剂之间的相互作用,还含有溶质一溶质分子之间的相互作用。对于固体溶质,还需要考虑其相变过程,这也是目前计算与预测分子溶解度所面临的挑战与难题之一。目前分子溶解度的计算方法对于结构比较简单的有机化合物效果则较好,但对于结构比较繁杂的、含有多个官能团的分子化合物则较为不适用,预测的效果也不是很好。不过这一问题对有机化合物脂水分配系数的计算实际上在一定程度上也存在这一现象。前文我们曾经采用量子化学密度泛函理论的方法,利用计算机和高斯软件对法匹拉韦及其衍生物的碱式电离常数pKb进行了预测,所得结果较好。本文基于密度泛函理论的B3LYP法在6-31+G(d,p)水平下对取代法匹拉韦分子进行结构全优化,将优化得到的量子化学参数和ACD Lab 6.0预测pKb值建立线性回归模型,选择相关陛最好的回归方程,用其预测法匹拉韦及其衍生物的logS值(图1),这对于了解法匹拉韦及其衍生物的药理及毒理质和新药研发有及其重要的意义。
1 计算方法
利用PC机及其软件Gauss View 5.0.9、Gaussian09W、Origin 7.5、ACD-Labs 6.0、Chemdraw 8.0等软件);利用Gaussion 09 W软件包,构建20个用ACD-Lab 6.0预测得到pKb的法匹拉韦衍生物的分子结构模型,然后用Gaussion View 5.09软件在DFT/B3LYP/6-31+G(d,p),POP=(muHiken,nbo,chelpg,hirshfeld)水平和条件下,对20种法匹拉韦衍生物进行结构优化,计算得到相应的量化参数,记录11-0的MiHiken电荷值和自然原子电荷轨道(NBO)值以及静电势电荷(ESP);用Mulliken、NBO和ESP电荷值(横坐标)分别对ACD Lab 6.0预测得到的logS(纵坐标)用Origin7.5作图,得到四个线性回归方程,得出相关系数,并比较各相关系数的大小,R值越接近1或-1,则相关性就越好,反之,相关性就越差。筛选出R值较大的线性方程作为预测方程。选取10种未知水溶液溶解度logS的法匹拉韦衍生物,用该量化方程计算其logS值,并与目前最流行的ACD Lab 6.0软件直接计算的得到的logS比较,验证密度泛函理论方法预测法匹拉韦及其衍生物分子的水溶解度值的准确性(图1)。
2 结果和讨论
2.1 不同基组对法匹拉韦氧原子电荷的影响
为测试基组对于法匹拉韦及其衍生物的11号氧原子四种电荷适应性与依赖性,首先使用流行的10种基组,包括pople's 6-31G,6-31G*,6-31G**,6-31++G**,6-311++G**和dunning's的相关一致性基组(CC-pVDZ,CC-pVTZ和Aug-CC-pVTZ)。结果表明,选择不同的基组对计算氧原子ESP、HIR和NBO电荷的影响不太显著(相对标准偏差std≤±1.7%),但是它们却对体系密立根电荷MUL值影响甚大(std=±8.7%)(表1)。
2.2 不同泛函对法匹拉韦氧原子电荷的影响
固定6-31+g(d,p)基组,选用经常采用的近似10种泛函如beckhandh,BLYP,B3LYP,beck-97ggal,MPWIK,becke98,HCTH407p,TPSS和M06-2X等,它们基本覆盖了最流行的GGA,metagga和混合交换相关势的形式。测试并计算了法匹拉韦氧原子电荷值的泛函依赖性。结果如表2所示。选择不同的泛函对计算法匹拉韦氧原子电荷的影响,除密立根电荷(MUL)外,对其余三种电荷(NBO,ESP,HIR)的影响也是不太显著(std≤±2.0%)。结合表1和表2数据可以看出,应用分子中的原子理论(AIM)方法,采用不同的基组和泛函组合,除密立根电荷值外,其余三种电荷值,于法匹拉韦及其衍生物系统氧原子电荷的计算,与实验值定性符合,不会产生显著的依赖性(低于±2%)。
2.3 羰基氧上的四种类型电荷值比较
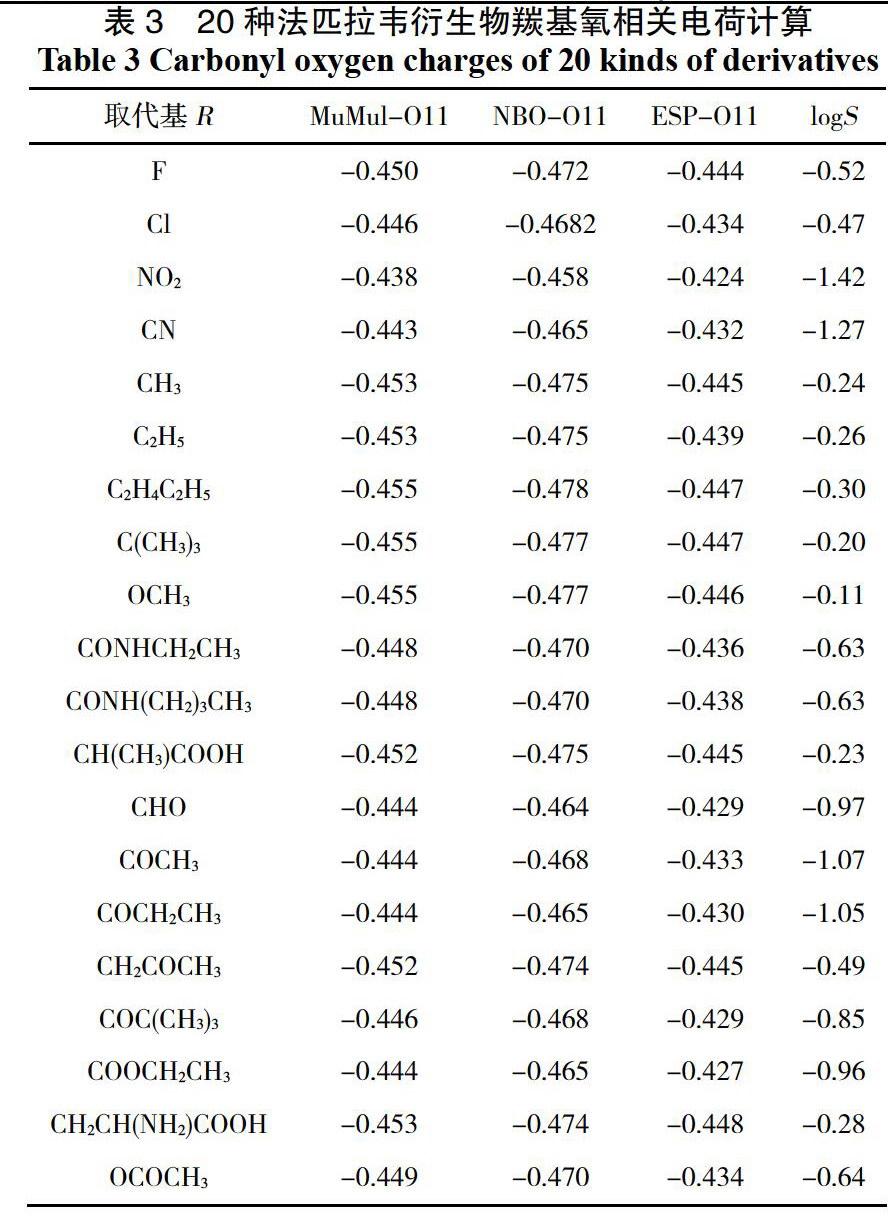
本次实验中,模拟所得的20种未知logS值的法匹拉韦衍生物的羰基氧上的Mulliken、NBO和ESP电荷值以及相对应的由ACD Lab6.0软件预测得知的logS如表3所示。
分别将羰基氧的MUL、NBO、HIR和ESP电荷值与相对应的log5作图,得到图2-4(省去图3,4),可以看出相关系数R,分别为0.716,0.956,0.902和0.986,发现ESP-011电荷的相关性最好(0.986,相关系数R越大越好),所以我们将采用此线性方程来进行预测后面的法匹拉韦衍生物的溶解度值(5,单位mol·L-1)。
2.4 羰基氧上的ESP电荷值与谁溶解度相关性
将所选的11种未知溶解度值的法匹拉韋的衍生物用opt进行结构优化,用DFT/B3LYP方法,采用6-31+G(d,p)基组进行分子计算,将得到的羰基氧的ESP-O电荷数据分别带入选定的线性回归方程中,计算得到相应化合物的logS值,拟合结果见表4。预测得到的logS值和ACD-Labs 6.0预测的logS值十分接近,最大偏差绝对对数值仅为0.08。这与其他理论模型方法预测的有机分子水溶解度对数值误差在一个单位值,效果较好。
图2(a)是法匹拉韦分子的静电势电荷分布图,浅红色氧原子部分,显示其氧原子带有较强的电负性。图2(b)是甲基取代的法匹拉韦衍生物分子的静电势电荷分布图,深红色氧原子部分,显示其氧原子带有强的电负性。
3 结论
(1)基于分子中的原子理论(AIM)框架下的-电荷分解法,测试了不同的基组与泛函对法匹拉韦药物分子氧原子的电荷的影响。结果显示,除了密立根电荷(MUL-0)外,不同的基组与泛函对其余三种氧原子电荷(NB0-0,ESP-0,HIR-0)拟合的值变化不大,影响也不显著;
(2)运用Gaussion 09W和Gaussion View 5.09软件,采用密度泛函活性方法,DFT B3LYP和6-31+G(d,p)基组,优化了20种法匹拉韦的分子结构,发现羰基氧上的ESP电荷值与log5值的相关性要好于NBO电荷值和静电势电荷ESP,相关系数达0.986,由此可以得出,法匹拉韦发挥其药理和毒理作用的部位很可能是在羰基的氧上。
(3)用上述的计算方法,我们计算了12种未知logS值的法匹拉韦衍生物(包括法匹拉韦)的ESP电荷值,并将其代入拟合所得较佳的线性方程,预测得到的logS值和ACD-Labs 6.0预测的logS值十分接近,最大偏差绝对对数值仅为0.18。发展基于第一性原理的高精度能量理论是今后一段时期理论工作者努力的方向。我们期待出现更为有效的预测多官能团分子溶解度的理论方法。
