
UPLC法同时测定缬沙坦氢氯噻嗪片中缬沙坦和氢氯噻嗪的含量
2017-07-07 15:09:13刘清华北京市药品检验所生命园实验室北京102206
中国药房 2017年15期
纪 宏,刘 晶,王 威,祁 进,刘清华(北京市药品检验所生命园实验室,北京 102206)
UPLC法同时测定缬沙坦氢氯噻嗪片中缬沙坦和氢氯噻嗪的含量
纪 宏*,刘 晶,王 威#,祁 进,刘清华(北京市药品检验所生命园实验室,北京 102206)
目的:建立同时测定缬沙坦氢氯噻嗪片中缬沙坦和氢氯噻嗪含量的方法。方法:采用超高效液相色谱法。色谱柱为Phenomenex C18,流动相为[0.1%磷酸溶液-乙腈(95∶5,V/V)]-[0.1%磷酸溶液-乙腈(5∶95,V/V)](梯度洗脱),流速为0.25m L/m in,检测波长为272 nm,柱温为35℃,进样量为1.5μL。结果:缬沙坦和氢氯噻嗪检测质量浓度线性范围分别为8.1~324.2μg/m L(r=0.999 9)、1.2~50.1μg/m L(r=0.999 9);定量限分别为0.24、0.04 ng,检测限分别为0.06、0.01 ng;精密度、稳定性、重复性试验的RSD<2.0%;加样回收率分别为97.69%~100.35%(RSD=1.03%,n=9)、98.27%~100.60%(RSD=0.83%,n=9)。结论:该方法操作简单、快速,结果准确,可用于缬沙坦氢氯噻嗪片中缬沙坦和氢氯噻嗪含量的同时测定。
超高效液相色谱法;缬沙坦氢氯噻嗪片;缬沙坦;氢氯噻嗪;含量
缬沙坦氢氯噻嗪片(Co-Diovan)是由缬沙坦和氢氯噻嗪组成的复方制剂,为瑞士诺华公司研发。该制剂的主要成分缬沙坦属血管紧张素Ⅱ受体拮抗药[1-2],辅助成分氢氯噻嗪则为利尿药[1-3],二者可协同治疗单一药物不能有效控制的轻、中度原发性高血压[3-4]。该制剂在强效降压的同时还具有保护心、脑、肾等靶器官的作用[1-4]。目前,2015年版《中国药典》[5]仅收载了测定缬沙坦、氢氯噻嗪单一成分的制剂,未收载复方制剂;且现有文献[6-10]测定缬沙坦氢氯噻嗪复方制剂含量的方法主要是高效液相色谱法,操作较为烦琐、耗时。因此,为更快速、准确地测定该复方制剂中缬沙坦和氢氯噻嗪的含量,笔者采用超高效液相色谱法(UPLC)建立了同时测定其中上述两种成分含量的方法。
1 材料
1.1 仪器
Acquity型UPLC仪,包括二元高压梯度混合系统、标准型自动进样器、柱温箱、二极管阵列检测器和Empower色谱工作站(美国Waters公司);XT105型电子天平(瑞士Mettler-Toledo公司);SB-25-12D型超声波清洗机(宁波新芝生物科技股份有限公司,功率:250W,频率:40 kHz);培英DZ-900型振荡器(太仓市实验设备厂)。
1.2 药品与试剂
缬沙坦氢氯噻嗪片(某企业,批号:T7038、T7039、T7017,规格:每片含缬沙坦80mg、氢氯噻嗪12.5mg);缬沙坦对照品(欧洲药典委员会,批号:H3409,纯度:99.0%);氢氯噻嗪对照品(欧洲药典委员会,批号:H3020,纯度:99.9%);乙腈为色谱纯,其余试剂均为分析纯,水为超纯水。
2 方法与结果
2.1 色谱条件
色谱柱:Phenomenex C18(50mm×2.1mm,1.7μm);流动相:[0.1%磷酸溶液-乙腈(95∶5,V/V)](A)-[0.1%磷酸溶液-乙腈(5∶95,V/V)](B),采用梯度洗脱(洗脱程序见表1);流速:0.25m L/m in;检测波长:272 nm;柱温:35℃;进样量:1.5μL。
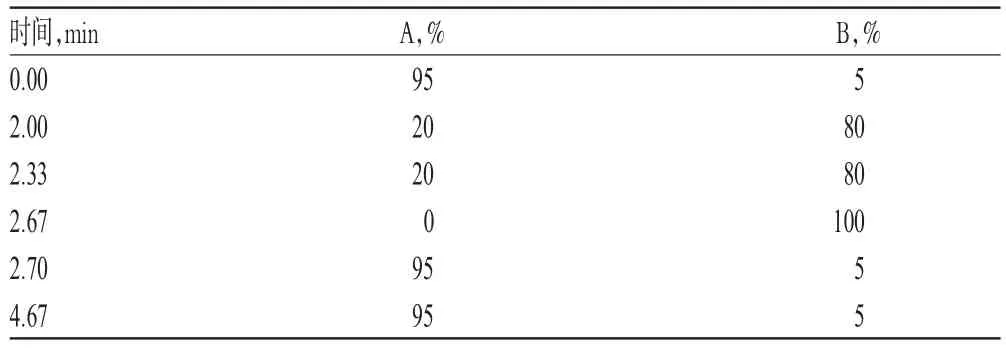
表1 梯度洗脱程序Tab 1 Gradientelution procedure
2.2 溶液的制备
2.2.1 混合对照品溶液 精密称取缬沙坦和氢氯噻嗪对照品各适量,加50%乙腈溶液溶解制成缬沙坦、氢氯噻嗪质量浓度分别为810.5、125.2μg/m L的单一对照品溶液。取上述单一对照品溶液各适量,置于同一50m L量瓶中,加50%乙腈溶液溶解制成缬沙坦、氢氯噻嗪质量浓度分别为8.00、1.25μg/m L的混合对照品溶液。
2.2.2 供试品溶液 取样品20片,精密称定,研细,精密称取适量(约相当于缬沙坦80mg),置于100m L量瓶中,加50%乙腈溶液80m L,超声处理25 m in,振摇40 min,放冷,加50%乙腈溶液稀释至刻度,摇匀,滤过,精密量取续滤液5m L,置于50m L量瓶中,加50%乙腈溶液稀释至刻度,摇匀,即得。
2.2.3 阴性对照溶液 按样品的处方比例和制备工艺制备缺缬沙坦和氢氯噻嗪的阴性样品,再按“2.2.2”项下方法制备阴性对照溶液。
2.3 系统适用性试验
精密量取“2.2”项下混合对照品溶液、供试品溶液和阴性对照溶液各适量,按“2.1”项下色谱条件进样测定,记录色谱,详见图1。由图1可知,在该色谱条件下,各成分均能达到基线分离,分离度>2.0;理论板数以缬沙坦峰计为10 000,以氢氯噻嗪峰计为35 000,保留时间分别为2.11、1.12 min。结果表明,其他成分对测定不干扰。
2.4 线性关系考察
分别精密量取“2.2.1”项下缬沙坦、氢氯噻嗪单一对照品溶液各0.1、0.2、0.6、1.0、1.4、2.0、4.0m L,置于同一10m L量瓶中,加50%乙腈溶液定容,摇匀,制成系列混合对照品溶液。精密量取上述系列混合对照品溶液各1.5μL,按“2.1”项下色谱条件进样测定,记录峰面积。以缬沙坦和氢氯噻嗪质量浓度(x,μg/m L)为横坐标、峰面积(y)为纵坐标进行线性回归,得二者回归方程分别为y=4 537 410x+5 789.3(r=0.999 9)、y= 23 498 311x-641.40(r=0.999 9)。结果表明,缬沙坦和氢氯噻嗪检测质量浓度线性范围分别为8.1~324.2、1.2~50.1μg/m L。
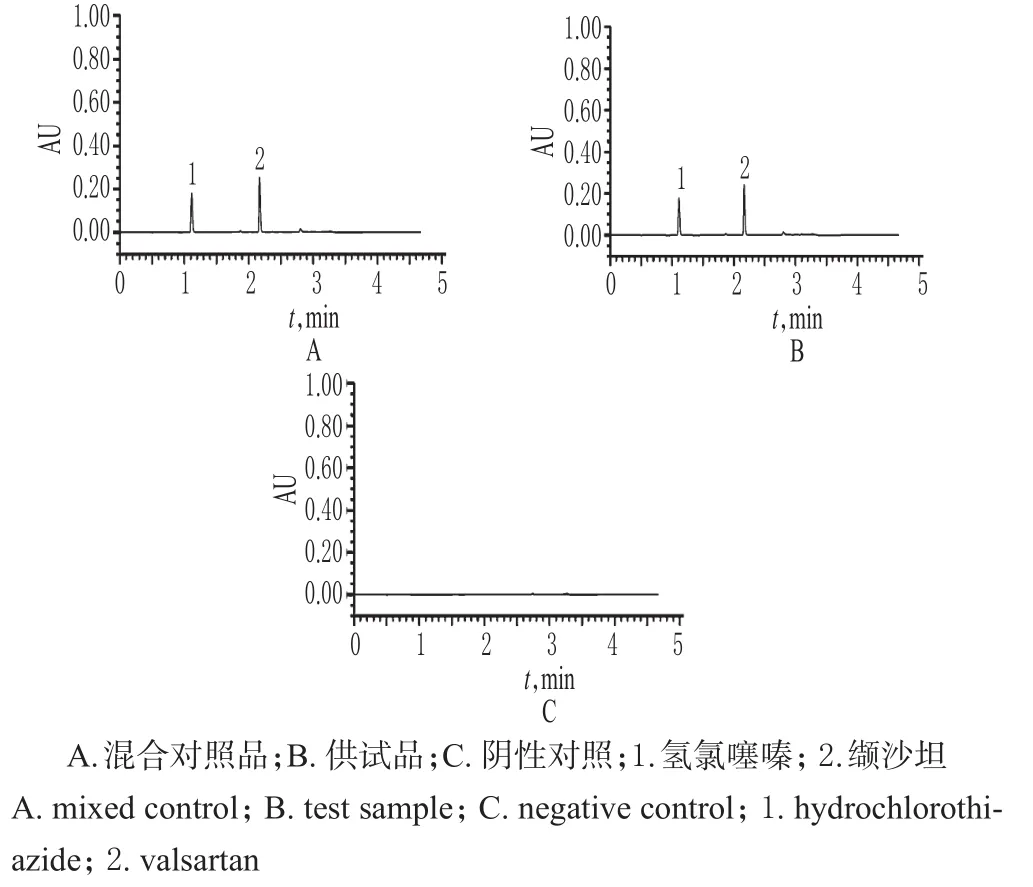
图1 超高效液相色谱图Fig 1 UPLC chromatograms
2.5 定量限与检测限考察
取“2.2.1”项下混合对照品溶液适量,倍比稀释,按“2.1”项下色谱条件连续进样测定6次,记录峰面积。当信噪比为10∶1时,得定量限(LOQ);当信噪比为3∶1时,得检测限(LOD)。结果,缬沙坦和氢氯噻嗪的LOQ分别为0.24、0.04 ng,LOD分别为0.06、0.01 ng。
2.6 精密度试验
取“2.2.1”项下混合对照品溶液适量,按“2.1”项下色谱条件连续进样测定6次,记录峰面积。结果,缬沙坦和氢氯噻嗪峰面积的RSD分别为0.27%、0.29%(n=6),表明仪器精密度良好。
2.7 稳定性试验
精密量取同一供试品溶液(批号:T7038)适量,分别于室温下放置0、2、4、8、12 h时按“2.1”项下色谱条件进样测定,记录峰面积。结果,缬沙坦和氢氯噻嗪峰面积的RSD分别为0.42%、0.67%(n=5),表明供试品溶液在室温下放置12 h内稳定性良好。
2.8 重复性试验
取样品(批号:T7038)适量,共6份,按“2.2.2”项下方法制备供试品溶液,再按“2.1”项下色谱条件进样测定,记录峰面积并计算含量。结果,缬沙坦和氢氯噻嗪的平均含量分别为95.30%和97.69%,RSD分别为0.97%、1.06%(n=6),表明本方法重复性良好。
2.9 加样回收率试验
取已知含量的样品(批号:T7039)适量,共9份,各置于50m L量瓶中,分别加入低、中、高质量的缬沙坦、氢氯噻嗪对照品,按“2.2.2”项下方法制备供试品溶液,再按“2.1”项下色谱条件进样测定,记录峰面积并计算加样回收率,结果见表2。
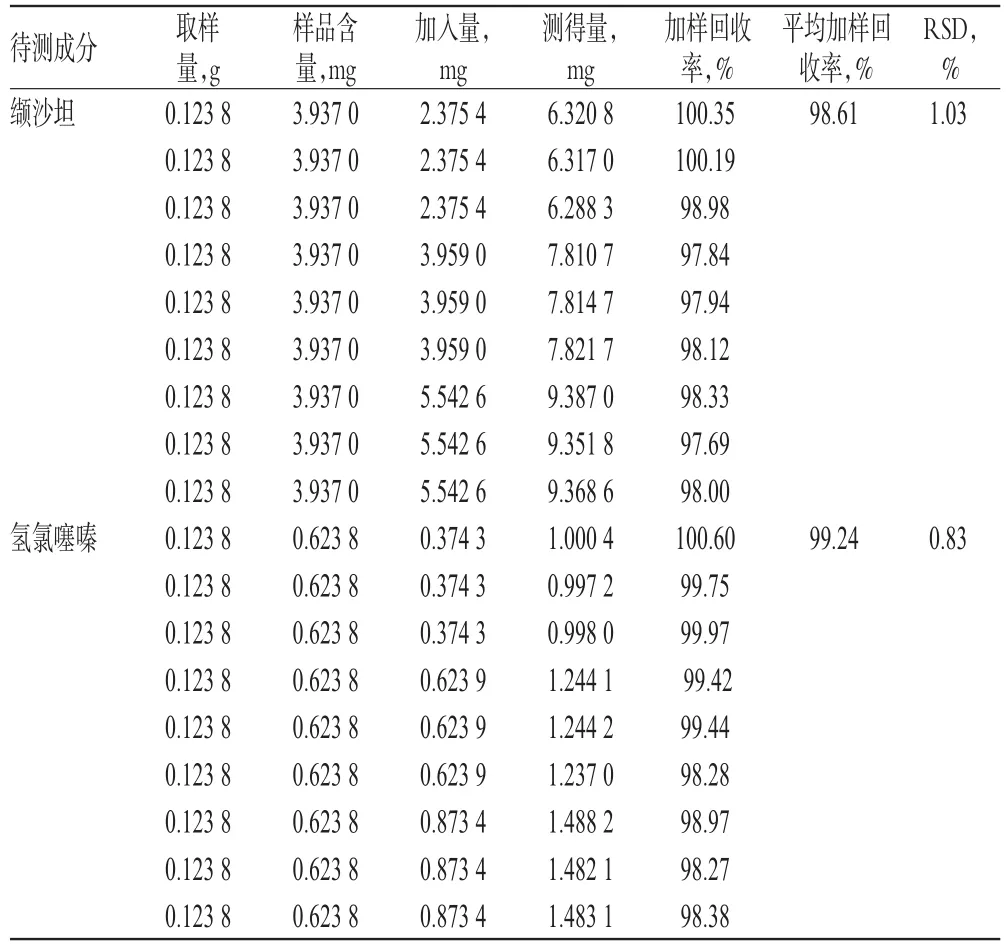
表2 加样回收率试验结果(n=9)Tab 2 Resultsof recovery tests(n=9)
2.10 样品含量测定
取3批样品各适量,按“2.2.2”项下方法制备供试品溶液,再按“2.1”项下色谱条件进样测定,记录峰面积并按外标法以峰面积计算缬沙坦和氢氯噻嗪的含量,结果见表3。

表3 样品含量测定结果(n=3,%)Tab 3 Results of content determination of samples(n=3,%)
3 讨论
3.1 检测波长的选择
分别取“2.2.1”项下缬沙坦、氢氯噻嗪的单一对照品溶液各适量,在210~400 nm波长范围内进行扫描。结果显示,缬沙坦在250 nm波长处、氢氯噻嗪在272 nm和323 nm波长处有较强吸收。因此,为使色谱基线平稳,干扰少,吸收峰更强,同时兼顾两种成分的测定,选择272 nm作为本试验的检测波长。
3.2 柱温的选择
本试验考察了不同柱温(15、25、35℃)对色谱峰分离的影响。结果,以35℃时的分离度最好,洗脱时间最短。因此,综合考虑色谱柱的使用情况等因素,最终本试验将柱温定为35℃。
3.3 溶剂的选择
本试验的溶剂先后考察了甲醇、乙腈、流动相等,结果采用乙腈的溶解性最好,对色谱柱的损伤也较小,且相对节约成本;同时,还考察了不同体积分数(25%、50%、75%)乙腈的溶解效果,结果采用50%乙腈溶液为溶剂时样品可以溶解完全。因此,本试验最终选择50%乙腈溶液为溶剂。
综上所述,本方法操作简单、快速,结果准确,可用于缬沙坦氢氯噻嗪片中缬沙坦和氢氯噻嗪含量的同时测定。
[1] 李文举,李敏,张伟.血管紧张素转换酶抑制剂的临床应用进展[J].中国药房,2010,21(20):1918-1920.
[2] 张石革.抗高血压药发展新趋向:复方制剂的进展与临床评价[J].中国医院用药评价与分析,2011,11(2):102-104.
[3] 张石革,王文.抗高血压药联合治疗的进展与评价:心血管疾病合理用药专家圆桌会议纪要[J].中国医院用药评价与分析,2011,11(2):97-99.
[4] 杜海洲,刘娜,刘桂玲,等.国际抗高血压复方制剂研发进展[J].中国新药杂志,2010,19(18):1676-1679.
[5] 国家药典委员会.中华人民共和国药典:二部[S].2015年版.北京:中国医药科技出版社,2015:771、1548-1549.
[6] 吴善霞,刘延凤.HPLC法同时测定缬沙坦氢氯噻嗪片中两组分的含量[J].药学研究,2013,32(3):153-154.
[7] 吴迪,郭伟英.HPLC法同时测定缬沙坦氢氯噻嗪胶囊中缬沙坦和氢氯噻嗪的含量[J].中国药房,2012,23(9):846-848.
[8] 廖红娟,张涛.高效液相色谱法测定缬沙坦片的含量[J].中国药业,2010,19(9):36-38.
[9] 杨卿.高效液相色谱法测定复方缬沙坦片中缬沙坦的含量[J].山西医科大学学报,2009,40(12):1093-1095.
[10] 饶五湖,林瑞群.高效液相色谱法测定氢氯噻嗪片的含量[J].中国药业,2009,18(17):22-24.
(编辑:刘 柳)
Simultaneous Determ ination of Valsartan and Hydrochlorothiazide in Valsartan Hydrochlorothiazide Tablets by UPLC
JIHong,LIU Jing,WANGWei,QIJin,LIU Qinghua(Shengm ingyuan Laboratory,Beijing Institute for Drug Control,Beijing 102206,China)
OBJECTIVE:To establish amethod for simultaneous determ ination of valsartan and hydrochlorothiazide in Valsartan hydrochlorothiazide tablets.METHODS:UPLC was adopted.The determ ination was performed on Phenomenex C18column w ith mobile phase consisted of[0.1%phosphoric acid solution-acetonitrile(95∶5,V/V)]-[0.1%phosphoric acid solution-acetonitrile(5∶95,V/V)](gradient elution)at the flow rate of 0.25m L/min.The detection wavelength was set at 272 nm,and the column temperature was 35℃.The sample size was 1.5μL.RESULTS:The linear range were 8.1-324.2μg/m L for valsartan(r=0.999 9)and 1.2-50.1μg/m L for hydrochlorothiazide(r=0.999 9).The limits of quantitation were 0.24,0.04 ng,and the limits of detection were 0.06,0.01 ng.RSDs of precision,stability and reproducibility tests were less than 2.0%;recoveries were 97.69%-100.35%for valsartan(RSD=1.03%,n=9)and 98.27%-100.60%for hydrochlorothiazide(RSD=0.83%,n=9).CONCLUSIONS:Themethod is simple,rapid and accurate,and can be used for the simultaneous determination of valsartan and hydrochlorothiazide in Valsartan hydrochlorothiazide tablets.
UPLC;Valsartan hydrochlorothiazide tablet;Valsartan;Hydrochlorothiazide;Content
R927.2
A
1001-0408(2017)15-2131-03
2016-05-31
2017-03-01)
*副主任药师,硕士。研究方向:药物质量控制分析。电话:010-51159668。E-mail:jihong@bidc.org.cn
#通信作者:副主任药师,硕士。研究方向:药物质量控制分析。电话:010-51159668。E-mail:wangwei@bidc.org.cn
DOI 10.6039/j.issn.1001-0408.2017.15.32
猜你喜欢
世界最新医学信息文摘(2021年12期)2021-06-09 08:37:02
中华养生保健(2020年10期)2021-01-18 06:46:08
中华养生保健(2020年5期)2020-11-16 01:44:34
实用中西医结合临床(2015年7期)2015-02-28 16:30:39
湖北理工学院学报(2015年1期)2015-02-27 15:02:36
卫生职业教育(2014年16期)2014-05-16 03:48:28
精细石油化工(2014年3期)2014-03-14 03:01:12
中医研究(2014年2期)2014-03-11 20:28:18
河南医学研究(2014年1期)2014-02-27 14:51:16
