
利用GBS技术研究240份宽皮柑橘的系统演化
2017-06-05 14:16:19王小柯江东孙珍珠
中国农业科学 2017年9期
王小柯,江东,2,孙珍珠
(1西南大学柑橘研究所,重庆 400712;2中国农业科学院柑橘研究所,重庆 400712)
利用GBS技术研究240份宽皮柑橘的系统演化
王小柯1,江东1,2,孙珍珠1
(1西南大学柑橘研究所,重庆 400712;2中国农业科学院柑橘研究所,重庆 400712)
【目的】GBS(genotyping-by-sequencing)是一种高效而经济的SNP(单核苷酸多态性)发掘和基因分型技术。采用GBS技术对240份宽皮柑橘进行基因分型,以阐明一些野生宽皮柑橘和地方品种的遗传背景,为其起源和演化研究提供更可靠的证据。【方法】选用国家柑橘种质重庆资源圃保存的具有广泛遗传多样性和地理起源的240份宽皮柑橘作为材料,利用EcoR I限制性内切酶消化基因组DNA后构建GBS文库;然后进行Illumina HiSeq PE150二代测序获得短读序列,通过BWA软件将序列映射到克里曼丁参考基因组上,再利用SAMTOOLS软件鉴定SNP位点。依据SNP的基因分型结果,采用邻近法构建系统演化树,并进行主成分分析。【结果】利用GBS简化基因组测序技术对240份宽皮柑橘进行测序,共获得96.3 Gb的测序数据,平均每个样本测序数据为401.26 Mb,经过测序深度为4X、Miss0.2、次要等位基因频率(MAF)>0.01的筛选条件过滤,最后共获得了114 200个高质量的SNP位点。主成分分析结果显示240份宽皮柑橘被分为4大类,其中温州蜜柑亚群、野生宽皮柑橘亚群可明显区分于其他宽皮柑橘。利用系统演化树可将240份宽皮柑橘划分到11个类群中。系统演化树和主成分分析都揭示了不同地理来源和特定形态的宽皮柑橘在遗传水平上存在明显的差异,比如来源于日本的温州蜜柑、欧美的克里曼丁橘及其杂种后代,以及中国南方的野生宽皮柑橘由于地理分布不同而形成了较为独特的类型,彼此间能够相互区分开。进化树结果表明中国南、北不同地域的宽皮柑橘可能存在不同的演化路径,南岭山脉及南方地区的野生宽皮柑橘、酸橘和目前南方地区栽培的砂糖橘存在较近的起源演化联系,而北方宽皮柑橘的演化却与宽皮柑橘中的古老地方品种存在紧密联系。人工杂交育种、长期的人工选择和驯化形成了不同类型的宽皮柑橘,同时也导致宽皮柑橘遗传多样性的增加。另外,一些宽皮柑橘资源中的可疑亲本也通过GBS技术得以准确鉴定。本研究表明沃柑与金诺橘有较近的亲缘关系。【结论】GBS技术用于柑橘种质资源的基因分型高效可靠,建立的系统演化树可以对240份宽皮柑橘进行准确划分,与用植物形态学划分的结果高度吻合。另外,GBS技术用于资源材料的准确鉴定和亲缘关系的研究,可为柑橘植物新品种权的保护提供可靠的技术支撑。
GBS;宽皮柑橘;系统演化;品种鉴定
0 引言
【研究意义】中国是世界宽皮柑橘的起源中心,宽皮柑橘种类丰富多样,至今仍有野生宽皮柑橘的分布[1],这为研究宽皮柑橘的起源和演化提供了重要材料。现有的研究表明宽皮柑橘是柑橘属下的基本种之一,但由于宽皮柑橘与柑橘属下的其他种易发生种间杂交,导致其种类繁多,不仅增加了鉴定难度,其亚种或类群的划分一直以来也存在较大的争议[2]。通过基因分型对宽皮柑橘种质进行甄别和鉴定不仅是资源收集、保存、评价和利用的基本要求,也为宽皮柑橘品种类群的划分、起源演化的研究、柑橘植物新品种权的保护和认定提供更加科学、充分的依据。在长期的栽培、驯化历史过程中,来源于不同地域的宽皮柑橘可能保留了明显的具有地域特征的遗传差异。因此,对不同地理来源的宽皮柑橘进行遗传多样性研究,有助于了解这些材料的遗传背景,从而为宽皮柑橘的育种提供可利用的基因资源。【前人研究进展】目前对宽皮柑橘种质的鉴定除利用植物形态学特征进行分辨外,也采用分子标记技术[3-4]和基因测序技术[5]。储春荣等[3]利用SSR、Indel等分子标记证实了苏州地区的黄皮橘、朱红橘是两类重要且较为古老的橘类资源,长江上游流域的红橘是与朱红橘不同的另一古老橘类品种;高恒锦等[4]利用类似的方法证实了起源于中国南岭山脉的野生宽皮柑橘是一类独特的原始野生宽皮柑橘类型,表明原始野生柑橘可能是宽皮柑橘中的初生种。WU等[5]通过对柑橘全基因组进行比较分析后,发现克里曼丁的基因组中有柚类基因的渗入,而来自中国的 1份野生宽皮柑橘与其他宽皮柑橘存在明显的遗传差异。近来随着高通量测序技术的发展,基因分型的成本持续降低,GBS技术作为第二代深度测序基础上发展起来的简化基因组测序技术,通过采用酶切加标签的方法,使多样本高通量平行测序得以实现[6-7]。这不仅大大降低了基因测序的成本,也使对大样本全基因组的基因分型成为可能,对深入了解种质资源的遗传背景和系统演化具有重要意义[8-10]。同时GBS获得的短读序列可通过有参或无参基因组的形式进行拼接组装,进而获得高密度的SNP标记,利用这些SNP标记或者开发的bin标记可进行遗传图谱构建[11-12]、遗传图谱加密[13-15]、全基因组关联分析(GWAS)[16-18]及基因组的辅助组装等研究。目前GBS技术作为基因分型的重要手段,已经在遗传图谱构建[11-12,19-20]、遗传选择[10]、基因多样性研究[21-23]、种质鉴定[24-25]及品种识别[26-27]等领域得到广泛的应用。【本研究切入点】宽皮柑橘中的古老地方品种、野生资源在中国极其丰富,但国外对这些资源的研究很少涉及,因此,中国的野生和地方宽皮柑橘品种的遗传多样性值得深入研究。基于GBS技术的低成本、高通量和不依赖参考基因组等特点[28],将其用于宽皮柑橘种质资源的基因分型是可行的。【拟解决的关键问题】利用 GBS技术对240份宽皮柑橘的野生资源、地方品种以及国内外培育的杂种材料进行基因分型,通过系统演化和遗传多样性分析,以阐明这些宽皮柑橘的遗传背景及起源和演化。
1 材料与方法
试验于2016年在中国农业科学院柑橘研究所进行。
1.1 试验材料
本研究采用的240份宽皮柑橘材料均采自国家果树种质重庆柑橘资源圃,这些宽皮柑橘种质来自于不同时期、不同地区的资源收集活动,包括起源于中国南方地区的野生宽皮柑橘15份,地方品种77份,选育品种140份,遗传材料8份。在选育品种中包括有清见杂种材料11份,爱媛30号及爱媛28号杂种材料19份。这240份宽皮柑橘材料中来源于日本的温州蜜柑有40份,来源于欧美的宽皮柑橘材料32份,尼泊尔材料5份,其余为国内材料,具体材料名单见电子版附表1。
1.2 试验方法
1.2.1 DNA提取和GBS文库构建 2016年春季采集所有宽皮柑橘资源的嫩叶,利用植物基因组DNA提取试剂盒(Magen HiPure Plant DNA Mini Kit,Guangzhou,China)提取DNA。提取的样品DNA送诺禾致源进行DNA质检、建库和测序,利用Qubit® 2.0荧光测定计(Invitrogen,Carlsbad,USA)检测核酸浓度,同时在1%的琼脂糖凝胶上100 V电泳40 min检测DNA纯度,高质量的DNA用于GBS文库构建和测序。每个样本取1.5 μg DNA用于文库构建,首先应用限制性内切酶EcoR I对基因组进行酶切,酶切后的片段两端利用T4 DNA连接酶(NEB)加上适配体接头,对每个样品进行扩增,然后对样品进行混合,电泳回收350—400 bp区间的DNA条带,割取的片段进行纯化,纯化后产物用于测序,测序反应在Illumina HiSeq测序平台上进行双末端150 bp的测序。GBS分析的重复性、可靠性检测使用电子版附表1中56号(1-1)、57号(专有橘)和58号(1-2)3份种质材料作为生物学重复。
1.2.2 单核苷酸多态性(SNP)鉴定 每条测序数据按照条码划分到对应的样本中,对每条序列进行严格的质控筛选,去除原始测序序列中的接头序列和低质量的短读序列,包括单端测序中含有的N数量超过该条序列长度的10%,或者单端测序中低质量(Q≤5)碱基数超过该条序列长度50%的序列。剩下的高质量序列采用BWA(Burrows-Wheeler Aligner)程序将其匹配到克里曼丁参考基因组上(ftp://ftp.ncbi.nlm.nih. gov/genomes/all/GCF_000493195.1_Citrus_clementina_ v1.0)。SNP检测采用Samtools软件进行,对齐的匹配文件用Samtools软件转换为BAM文件后进行变异调用,Samtools收集BAM文件中的汇总信息,计算可能基因型的似然值,再利用Bcftools应用先验值进行变异调用,采用贝叶斯模型检测群体中的多态性位点获得VCFs文件。
1.2.3 群体进化树分析 采用SNP基因分型数据估测240份宽皮柑橘的系统演化,根据群体遗传学特征的共同点或差异推断出它们的亲缘关系远近。利用个体SNPs的检测值计算群体间的遗传距离。两个个体i和j之间的p-距离通过如下公式计算:
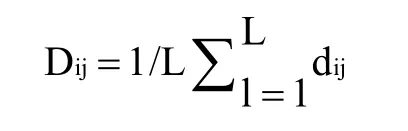
式中,L表示高质量SNPs区域长度,dij表示两个个体为不同基因型值的赋值表。运用 TreeBest(http:// treesoft.sourceforge.net/treebest.shtml)软件计算距离矩阵,以此为基础,通过邻接法(neighbor-joining method)构建系统进化树。引导值(bootstrap values)经过达1 000次计算获得。
1.2.4 主成分分析 通过GCTA(http://cnsgenomics. com/software/gcta/pca.html)软件计算特征向量以及特征值,并利用R软件绘制PCA分布图。
2 结果
2.1 测序质量
GBS测序得到的原始图像数据文件经碱基识别分析转化为原始测序序列,240个宽皮柑橘样本的总测序数据量为96.31 Gb,去除低质量的序列后,产生的高质量序列数据量为96.3 Gb,平均每个样本数据量为401.26 Mb。测序质量较高(Q20≥93.78%、Q30≥85.19%),GC分布正常,群体样本与克里曼丁参考基因组的平均比对率为93.79%,表明本试验的样本与参考基因组克里曼丁有较高的相似性。序列平均覆盖度为17.82%(至少有一个碱基的覆盖),至少有4个碱基的平均覆盖度为8.60%。利用Samtools软件检测后共获得765 252个SNP位点,经过条件为测序深度4×、Miss0.2、次要等位基因频率(MAF)>0.01的过滤后,最后共获得114 200个高质量的SNP位点用于群体进化树分析。
2.2 主成分分析
本研究选择的 240份宽皮柑橘涵盖了宽皮柑橘中的大部分类型,利用PCA分析方法对240份宽皮柑橘材料的亲缘关系进行了分析,根据第一主成分和第二主成分可将240份宽皮柑橘分为4个亚群,分别为类群Ⅰ(温州蜜柑亚群,包括温州蜜柑、早熟蜜橘和诺瓦橘),类群Ⅱ(野生宽皮柑橘亚群,包括细皮狗屎柑、大坑野橘、聂都野橘、莽山橘、粗皮狗屎柑、道县野橘),类群Ⅲ(包括大浦、早香和橘橙22-53),类群Ⅳ(其余宽皮柑橘及其杂种品种,包括酸橘、年橘、椪柑、红橘、朱红橘、地中海橘、早橘、橘橙或橘柚杂种、清见杂种、克里曼丁橘等)。具体结果见图1。

图1 240份宽皮柑橘的主成分分析图Fig. 1 The principal component analysis of 240 mandarins
2.3 群体进化树分析
群体进化树的结果表明,240份宽皮柑橘种质聚为一个大类,支持宽皮柑橘是柑橘属下一个基本种的说法。来源于不同地理区域的宽皮柑橘在遗传上存在明显的差异,比如来源于日本的温州蜜柑、来源于欧美的克里曼丁橘及其杂种与中国的绝大部分宽皮柑橘品种间存在明显的遗传差异,表明地理来源的不同可能是引起宽皮柑橘遗传差异的重要原因之一。根据进化树对240份宽皮柑橘进一步细分后,可将这些材料划分到11个宽皮柑橘亚群中(具体分类亚群见电子版附表1),这11个亚群分别是野生宽皮柑橘及其衍化种(类群Ⅰ,包括小果类型的酸橘、粗皮狗屎柑、大坑野橘、道县野橘、莽山橘、聂都野橘、年橘、砂糖橘、八月橘、马水橘、青皮蜜橘等),古老栽培品种(类群Ⅱ,包括土橘、建柑、椪柑、红橘、地中海橘、槾橘等),朱红橘(类群Ⅲ,包括朱红橘、南橘、料红、南丰蜜橘等),早橘(类群Ⅳ,包括本地早、城固冰糖橘、旺苍皱皮柑、克里曼丁×本地早杂种等),欧美橘橙杂种(类群Ⅴ,包括王柑、默科特、恩科尔、丽红、金诺、无核沃柑等),橘柚杂种(类群Ⅵ,包括明尼奥拉、清峰、汉源黄果柑、湘慈43号等),清见×椪柑杂种群(类群Ⅶ),爱媛系列杂种(类群Ⅷ,包括爱媛28号杂种、爱媛30号杂种),克里曼丁橘类(类群Ⅸ,主要为克里曼丁橘及其杂种后代,如费尔柴尔德、弗来蒙特橘、秋辉、帕森橘等以及其他类型杂种),温州蜜柑(类群Ⅹ),清见杂种(类群Ⅺ,清见,清见与天草的杂交种,清见与明尼奥拉的杂交种)。进化树的分类结果与利用植物形态学分类的结果是高度吻合的。
2.3.1 系统演化 从群体进化树的结果来看,中国南、北不同地域的宽皮柑橘可能存在不同的演化路径。来源于南岭山脉及南方地区的野生宽皮柑橘、宽皮柑橘中小果类型的酸橘、华南地区广泛种植的砂糖橘、八月橘、马水橘、青皮蜜橘等存在较近的亲缘关系,这些材料在地理分布上也主要位于华南地区,而分布于长江下游流域的小果宽皮柑橘中的乳橘类型,如南丰蜜橘、东华蜜橘却与朱红橘有较近的亲缘关系,而小果类中的本地早却与古老宽皮柑橘中的旺苍皱皮柑有较近的亲缘关系。结合这些品种的地理分布,推测南方地区栽植的砂糖橘等类型可能是从小果类的野生宽皮柑橘演化为酸橘后,再由其演化为现代栽培宽皮柑橘品种。同时还发现南方地区的红橘和北方橘区的朱红橘之间也存在明显的差异,这表明即便都是红皮橘类型,南北地理气候差异也造成了两个不同的类群。另外从进化树上看,中国古老栽培品种旺苍皱皮柑与城固冰糖橘遗传距离较近,两者有明显的亲缘关系,并且可能是本地早的早期亲本来源之一,说明中国北方宽皮柑橘的演化有原始古老柑类品种的参与。来源于欧洲地中海地区的柳叶橘(阿凡娜橘)也形成了古老栽培品种中较为明显的一个分支,与来源于东亚地区的土橘、椪柑、红橘、朱红橘等彼此分开,进一步证实了不同地理来源的品种间存在较明显的遗传差距。

图2 240份宽皮柑橘群体进化树Fig. 2 The phylogenetic tree of 240 mandarins
2.3.2 遗传多样性 从群体进化树的结果可发现,宽皮柑橘与柑橘属其他种的种间杂交,会造成遗传多样性的增加,进而形成独特的亚群。比如来源于欧美的王柑、默科特等品种形成一个独特的亚群,这些品种多数来源于橘橙品种间的天然杂种,由于具有橙类的遗传基因,因此这个亚群的品种多数具有果皮硬、成熟晚等特点。而一些遗传背景更为复杂的橘橙或橘柚杂种则形成了新的亚群,如清峰、湘慈43、汉源黄果柑等。通过人工杂交获得的杂种后代也会导致遗传多样性的增加,而人工选择的偏好性使具有相似特征的品种形成新的品种亚群,比如清见与不同椪柑品种的杂交后代形成了一个新的亚群(图2中的类群Ⅶ),这些材料在遗传距离上与椪柑相距较远;而爱媛 30号、爱媛28号人工杂交后代也形成了新的分支,但这些杂种后代与母本爱媛30号、爱媛28号的遗传距离相对较近。来源于欧美的克里曼丁橘及其杂种后代在进化树上也形成了一个明显的分支,表明克里曼丁橘应是一个遗传背景较为复杂的宽皮柑橘类型,其参与了许多现代宽皮柑橘品种的形成,如费尔柴尔德、弗来蒙特橘、福琼橘、凯旋柑、秋辉、苔丝、南香等品种中都具有该品种的基因渗入。
2.3.3 未知亲本材料的鉴定 GBS技术不仅对研究宽皮柑橘的亲缘关系具有重要价值,同时也为未知材料亲本的鉴定提供了重要线索。在本研究中加入的56号(1-1)和58号(1-2)两个生物学重复,其聚类结果显示两份重复材料均能聚在一起,表明通过 GBS得到的SNP数据和最后的分类结果均是稳定可靠的。并且56号和58号两份材料与爱媛28号和天草的遗传距离都很近,这两份材料本身来源于爱媛28号的杂交后代,而爱媛28号又是天草和南香的杂交后代,由此可见,利用GBS数据可以准确推导出这些材料的直接亲本。另外,本研究也对沃柑的亲本进行了探索,以往沃柑被认为是坦普尔和丹西红橘的杂种后代[29],但根据叶片、果实等形态的鉴定结果表明沃柑不像是从丹西红橘衍生而来。本研究利用GBS技术发现沃柑与金诺的亲缘关系十分密切,遗传距离远低于金诺与韦尔金的遗传距离,而金诺和韦尔金属于姊妹系,都是王柑和柳叶橘的杂交后代,因此,沃柑极有可能是王柑和柳叶橘的杂种后代,或者就是金诺的杂交后代。这与国外的报道是一致的,根据第12届国际柑橘学会会议报道,利用SSR技术也证明了沃柑是金诺的杂交后代[30]。
同时 GBS技术对了解种质材料的遗传背景具有重要意义。盛田温州蜜柑具有果皮极光滑、不易浮皮等特点,以往被认为是宫川温州蜜柑的早生芽变材料,但从本研究结果来看盛田温州蜜柑与宫川温州蜜柑的遗传距离较远,不像是宫川温州蜜柑的芽变材料,极有可能是种子的实生繁殖后代。而大浦温州蜜柑以往被认为是山崎早生温州蜜柑芽变而来,但研究结果表明本试验的大浦温州蜜柑材料应为杂种来源,其芽变来源的可能性较小。
3 讨论
中国是世界宽皮柑橘的起源演化中心,野生宽皮柑橘类型多样,道县野橘的发现表明南岭山脉是南方宽皮柑橘的起源中心[4,31],本研究结果进一步表明野生宽皮柑橘与南方地区的酸橘存在紧密联系,可能从其中演化出了酸橘,进而再演化出了砂糖橘、马水橘等现代栽培的宽皮柑橘品种。
宽皮柑橘由于栽培历史久远、地理分布广泛、种间易发生杂交,因而遗传类型极其多样,对其划分存在诸多争议。美国的斯文格认为宽皮柑橘仅仅是柑橘属下的一个基本种[32],日本的田中长三郎将宽皮柑橘对应的蜜柑区进行了较为细致的划分,包括5个亚区共 36个种[33],而中国的曾勉教授根据植物学特性结合地理分布将宽皮柑橘分为柑和橘两个亚区[34],认为柑亚区为杂种起源。现代分子生物学证据已经表明,宽皮柑橘是柑橘属下的一个基本种[35],本研究支持该结论。从240份宽皮柑橘的群体进化树分析来看,将宽皮柑橘划分为一个种是合理的,但如何对诸多的宽皮柑橘资源进行有效管理,开展更细致的划分也是必要的。本研究中的系统演化和主成分分析都表明,温州蜜柑、野生宽皮柑橘、椪柑、克里曼丁橘彼此间存在明显的遗传差异,温州蜜柑、克里曼丁橘是具有明显杂种来源的宽皮柑橘类型,而椪柑是一个栽培宽皮柑橘类型,斯文格将其作为宽皮柑橘的代表种是不恰当的,原产中国南方地区的原始野生宽皮柑橘才真正具有宽皮柑橘代表种的地位,其可能衍生出了酸橘,进而衍生出了现代栽培宽皮柑橘砂糖橘等类型。
利用 GBS技术对宽皮柑橘进行更细致的分类研究,与用植物形态学分类的结果是吻合的,因而传统的宽皮柑橘按照植物学特征进行分类划分具有一定的合理性[36],比如本研究中的酸橘、椪柑、红橘、朱红橘、地中海橘、克里曼丁橘等亚群都能准确地得以划分。但由于宽皮柑橘与橙类、柚类的种间杂交易于发生,新的种间杂交还将不断促进新类型的产生,因此,利用 GBS技术对柑橘资源进行基因分型有助于材料的准确划分和科学管理。
GBS研究结果还表明,地理隔离、杂交育种、人工选择是造成资源特异性和多样性的内在动力,来源于不同地理区域的资源材料往往会带有明显的遗传印记,这与育种习惯、人工选择的偏好性有一定关系。人工的长期栽培和选择使得一些品种群形成明显类群,如椪柑、红橘、朱红橘、地中海橘、克里曼丁橘等栽培品种都形成了明显的亚群。同时骨干亲本在育种中的应用也促进了品种群的形成,比如欧美地区常利用王柑、默科特等为骨干亲本开展杂交,其后代品种往往具有晚熟、皮较硬的特征;而东亚地区的日本、韩国等常采用清见和椪柑进行杂交,获得一些肉质细嫩化渣、高糖的品种,进而形成了有明显遗传特征的亚群。这些都在本研究中得到充分的证实。
4 结论
本研究利用GBS技术对240份宽皮柑橘资源进行了简化基因组的基因分型,建立的系统演化树可以准确地对240份宽皮柑橘进行划分,其结果与用植物形态学的划分结果存在高度吻合,表明GBS技术可高效用于资源材料的准确鉴定和亲缘关系的研究,可作为资源材料分子鉴定的重要手段。基于GBS技术具有高通量、低成本等特点,今后可用GBS技术建立涵盖所有在圃柑橘种质资源的简化基因组的基因分型数据,为今后品种的鉴别和品种权保护提供依据。
[1] 贺善文. 柑橘类种质资源中心问题的初步探讨. 园艺学报, 1979,6(1): 19-25. HE S W. A preminary study of the native citrus in central China. Acta Horticulturae Sinica, 1979, 6(1): 19-25. (in Chinese)
[2] 周志钦. 柑橘分类研究进展一文献综述. 园艺学报, 1993, 20(3): 243-250.
ZHOU Z Q. Advances in citrus taxonomy: Literature review. Acta Horticulturae Sinica, 1993, 20(3): 243-250. (in Chinese)
[3] 储春荣, 江东, 高恒锦, 陈绍彬, 周坤杰. 苏州地区宽皮柑橘遗传多样性分析. 中国南方果树, 2016, 45(3): 1-8. CHU C R, JIANG D, GAO H J, CHEN S B, ZHOU K J. Genetic diversity of Mandarin germplasm in Suzhou district. South China Fruits, 2016, 45(3): 1-8. (in Chinese)
[4] 高恒锦, 储春荣, 王小柯, 陈绍彬, 晏承泉, 闫树堂. 45份宽皮柑橘野生和地方资源遗传多样性分析. 中国南方果树, 2016, 45(2): 1-9.
GAO H J, CHU C R, WANG X K,CHEN S B, YAN C Q, YAN S T. The genetic diversity of landrace mandarin germplasms. South China Fruits, 2016, 45(2): 1-9. (in Chinese)
[5] WU G A, PROCHNIK S, JENKINS J, SALSE J, HELLSTEN U, MURAT F, PERRIER X, RUIZ M, SCALABRIN S, TEROL J, et al. Sequencing of diverse mandarin, pummelo and orange genomes reveals complex history of admixture during citrus domestication. Nature Biotechnology, 2014, 32(7): 656-662.
[6] ELSHIRE R J, GLAUBITZ J C, SUN Q, POLAND J A, KAWAMOTO K, BUCKLER E S, MITCHELL S E. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE, 2011, 6(5): e19379.
[7] SONAH H, BASTIEN M, IQUIRA E, TARDIVEL A, LEGARE G, BOYLE B, NORMANDEAU E, LAROCHE J, LAROSE S, JEAN M, BELZILE F. An improved genotyping by sequencing (GBS) approach offering increased versatility and efficiency of SNP discovery and genotyping. PLoS ONE, 2013, 8(1): e54603.
[8] GLAUBITZ J C, CASSTEVENS T M, LU F, HARRIMAN, J., ELSHIRE R J, SUN Q. TASSEL-GBS: A high capacity genotyping by sequencing analysis pipeline. PLoS ONE, 2014, 9(2): e90346.
[9] POLAND J A, RIFE T W. Genotyping-by-sequencing for plant breeding and genetics. Plant Genome, 2012, 5(3): 92-102.
[10] POLAND J, ENDELMAN J, DAWSON J, RUTKOSKI J, WU S Y, MANES Y, DREISIGACKER S, CROSSA J, SANCHEZ-VILLEDA H, SORRELLS M, JANNINK J L. Genomic selection in wheat breeding using genotyping-by-sequencing. Plant Genome, 2012, 5(3): 103-113.
[11] POLAND J A, BROWN P J, SORRELLS M E, JANNINK J L. Development of high-density genetic maps for barley and wheat using a novel two-enzyme genotyping-by-sequencing approach. PLoS ONE, 2012, 7(2): e32253.
[12] WARD J A, BHANGOO J, FERNANDEZ-FERNANDEZ F, MOORE P, SWANSON J D, VIOLA R, VELASCO R, BASSIL N, WEBER C A, SARGENT D J. Saturated linkage map construction in Rubus idaeus using genotyping by sequencing and genome-independent imputation. BMC Genomics, 2013, 14(1): 2.
[13] SPINDEL J, WRIGHT M, CHEN C, COBB J, GAGE J, HARRINGTON S, LORIEUX M, AHMADI N, MCCOUCH S. Bridging the genotyping gap: using genotyping by sequencing (GBS) to add high-density SNP markers and new value to traditional bi-parental mapping and breeding populations. Theoretical and Applied Genetics, 2013, 126(11): 2699-2716.
[14] LU F, ROMAY M C, GLAUBITZ J C, BRADBURY P J, ELSHIRE R J, WANG T, LI Y, LI Y, SEMAGN K, ZHANG X, HERNANDEZ A G, MIKEL M A, SOIFER I, BARAD O, BUCKLER E S. Highresolution genetic mapping of maize pan-genome sequence anchors. Nature Communications, 2015, 6: 6914.
[15] ZHOU Z, ZHANG C, ZHOU Y, HAO Z, WANG Z, ZENG X, DI H, LI M, ZHANG D, YONG H, ZHANG S, WENG J, LI X. Genetic dissection of maize plant architecture with an ultra-high density bin map based on recombinant inbred lines. BMC Genomics, 2016, 17: 178.
[16] ROMAY M C, MILLARD M J, GLAUBITZ J C, PEIFFER J A, SWARTS K L, CASSTEVENS T M, ELSHIRE R J, ACHARYA C B, MITCHELL S E, FLINT-GARCIA S A, MCMULLEN M D, HOLLAND J B, BUCKLER E S, GARDNER C A. Comprehensive genotyping of the USA national maize inbred seed bank. Genome Biology, 2013, 14(6): R55.
[17] MORRIS G P, RAMU P, DESHPANDE S P, HASH C T, SHAH T, UPADHYAYA H D, RIERA-LIZARAZU O, BROWN P J, ACHARYA C B, MITCHELL S E, HARRIMAN J, GLAUBITZ J C, BUCKLER E S, KRESOVICH S. Population genomic and genome-wide association studies of agroclimatic traits in sorghum. Proceedings of the National Academy of Sciences of the United States of America, 2013, 110(2): 453-458.
[18] LI X, LI X, FRIDMAN E, TESSO T T, YU J. Dissecting repulsion linkage in the dwarfing gene Dw3 region for sorghum plant height provides insights into heterosis. Proceedings of the National Academy of Sciences of the United States of America, 2015, 112(38): 11823-11828.
[19] GARDNER K M, BROWN P, COOKE T F, CANN S, COSTA F,BUSTAMANTE C, VELASCO R, TROGGIO M, MYLES S. Fast and cost-effective genetic mapping in apple using next-generation sequencing. G3-Genes Genomes Genetics, 2014, 4(9): 1681-1687.
[20] GUAJARDO V, SOLIS S, SAGREDO B, GAINZA F, MUNOZ C, GASIC K, HINRICHSEN P. Construction of high density sweet cherry (Prunus avium L.) linkage maps using microsatellite markers and SNPs detected by genotyping-by-sequencing (GBS). PLoS ONE, 2015, 10(5): e0127750.
[21] LIN M, CAI S, WANG S, LIU S, ZHANG G, BAI G. Genotypingby-sequencing (GBS) identified SNP tightly linked to QTL for pre-harvest sprouting resistance. Theoretical and Applied Genetics, 2015, 128(7): 1385-1395.
[22] LU F, LIPKA A E, GLAUBITZ J, ELSHIRE R, CHERNEY J H, CASLER M D, BUCKLER E S, COSTICH D E. Switchgrass genomic diversity, ploidy, and evolution: Novel insights from a network-based SNP discovery protocol. PLoS Genetics, 2013, 9(1): e1003215.
[23] BAJAJ D, DAS S, BADONI S, KUMAR V, SINGH M, BANSAL K C, TYAGI A K, PARIDA S K. Genome-wide high-throughput SNP discovery and genotyping for understanding natural (functional) allelic diversity and domestication patterns in wild chickpea. Scientific Reports, 2015, 5: 12468.
[24] WONG M M, GUJARIA-VERMA N, RAMSAY L, YUAN H Y, CARON C, DIAPARI M, VANDENBERG A, BETT K E. Classification and characterization of species within the genus lens using genotyping-by-sequencing (GBS). PLoS ONE, 2015, 10(3): e0122025.
[25] WU B, ZHONG G Y, YUE J Q, YANG R T, LI C, LI Y J, ZHONG Y, WANG X, JIANG B, ZENG J W, ZHANG L, YAN S T, BEI X J, ZHOU D G. Identification of pummelo cultivars by using a panel of 25 selected SNPs and 12 DNA segments. PLoS ONE, 2014, 9(4): e94506.
[26] LOMBARDI M, MATERNE M, COGAN N O, RODDA M, DAETWYLER H D, SLATER A T, FORSTER J W, KAUR S. Assessment of genetic variation within a global collection of lentil (Lens culinaris Medik.) cultivars and landraces using SNP markers. BMC Genetics, 2014, 15: 150.
[27] CABEZAS J A, IBANEZ J, LIJAVETZKY D, VELEZ D, BRAVO G, RODRIGUEZ V, CARRENO I, JERMAKOW A M, CARRENO J, RUIZ-GARCIA L, THOMAS M R, MARTINEZ-ZAPATER J M. A 48 SNP set for grapevine cultivar identification. BMC Plant Biology, 2011, 11: 153.
[28] 黎裕, 李英慧, 杨庆文, 张锦鹏, 张金梅, 邱丽娟. 基于基因组学的作物种质资源研究: 现状与展望. 中国农业科学, 2015, 48(17): 3333-3353.
LI Y, LI Y H, YANG Q W, ZHANG J P, ZHANG J M, QIU L J. Genomics-based crop germplasm research: Advances and perspectives. Scientia Agricultura Sinica, 2015, 48(17): 3333-3353. (in Chinese)
[29] 江东, 曹立. 晚熟高糖杂柑品种‘沃柑’在重庆的引种表现. 中国南方果树, 2011, 40(5): 33-34.
JIANG D, CAO L. The phenotype of ‘Or’ (late-maturing high-sugar hybrid) after cultivating in Chongqing. South China Fruits, 2011, 40(5): 33-34. (in Chinese)
[30] BARRY G H, JR F G G, CHEN C, ROOSE M L, FEDERICI C T, MCCOLLUM G T. Investigating the parentage of ‘orri’ and ‘fortune’mandarin hybrids. Acta Horticulturae, 2015, 36(1065): 449-456.
[31] 廖振坤, 张秋明, 刘卫国, 丁伟平, 张玲. 南岭山脉宽皮柑橘近缘野生种亲缘关系鉴定. 湖南农业大学学报(自然科学版), 2006, 32(4): 385-388.
LIAO Z K, ZHANG Q M, LIU W G, DING W P, ZHANG L. Identification of relative relationships of wild relatives of eucitrus originated from Nanling mountains by AFLP analysis. Journal of Hunan Agricultural University (Natural Sciences), 2006, 32(4): 385-388. (in Chinese)
[32] SWINGLE W T. The botany of Citrus and its wild relatives. Citrus Industry, 1967: 190-430.
[33] TANAKA T. Fundamental discussion of Citrus classification. Studia Citrologica, 1977, 14: 1-6.
[34] 曾勉. 对柑橘分类的认识体会和整理的意见. 中国果树, 1962(2): 31-37.
ZENG M. The understanding and opinion of the citrus classification. China Fruits, 1962(2): 31-37. (in Chinese)
[35] 谢让金, 周志钦, 邓烈. 真正柑橘果树类植物基于AFLP分子标记的分类与进化研究. 植物分类学报, 2008, 46(5): 682-691.
XIE R J, ZHOU Z Q, DENG L. Taxonomic and phylogenetic relationships among the genera of the True Citrus Fruit Trees Group (Aurantioideae, Rutaceae) based on AFLP markers. Journal of Systematics and Evolution (formerly Acta Phytotaxonomica Sinica), 2008, 46(5): 682-691. (in Chinese)
[36] 周开隆, 叶荫民. 中国果树志·柑橘卷. 北京: 中国林业出版社, 2010.
ZHOU K L, YE Y M. China Fruit’s Monograph: Citrus Volume. Beijing: China Forestry Publishing Press, 2010. (in Chinese)
(责任编辑 赵伶俐)
Study on Phylogeny of 240 Mandarin Accessions with Genotyping-by-Sequencing Technology
WANG XiaoKe1, JIANG Dong1,2, SUN ZhenZhu1
(1Citrus Research Institute, Southwest University, Chongqing 400712;2Citrus Research Institute of Chinese Academy of Agricultural Sciences, Chongqing 400712)
【Objective】Genotyping-by-sequencing (GBS) is an economic technique to discover SNP and genotype myriad of crop germplasms in an effective way. The aim of this study is to clarify the classification and evolution of 240 mandarin germplasms by using GBS. 【Method】 A total of 240 mandarin germplasms conserved in the National Citrus Germplasm Repository in Chongqing, with widely genetic diversity and geographic origin, were selected as trial materials. GBS library was constructed withgenomic DNAs after digested with EcoR I restriction endonuclease and sequenced on Illumina HiSeq PE150, then the sequences were mapped to the clementine (Citrus clementina hort. ex Tanaka) reference genome by using BWA, and SNPs were called with the SAMTOOLS pipeline. With the SNPs genotyping data, a phylogenetic tree was built by using Neighbor-joining method and a principal component analysis (PCA) was carried out. 【Result】By using GBS, a total of 96.3 Gb of sequences were generated from the 240 mandarin germplasms, and each sample produced 401.26 Mb in average. After screening with parameter of dp4, miss0.2 and minor alleles frequency (MAF)>0.01, a total of high quality 114 200 SNP sites were retained. The PCA analysis showed that the 240 mandarin germplasms could be divided into 4 groups, in which satsuma sub-group and wild mandarin sub-group could be clearly separated from other mandarin accessions. With phylogenetic analysis, the 240 mandarin germplasms could be divided into 11 groups. Both the phylogenetic analysis and PCA suggested that the genetic variations were presented in mandarin germplasms with different geographical origins and morphological characteristics. For example, satsuma mandarin (Citrus unshiu Macf.) derived from Japan, clementine (Citrus clementina Hort.ex. Tanaka) and its offspring from Europe and America, as well as wild mandarins from China could be clearly distinguished based on phylogenetic tree, moreover the phylogenetic tree showed that the mandarin germplasms derived from the Southern and Northern of China have unique evolutionary route. The wild mandarins distributed in Nanling Mountain and southern China have a closer phylogenetic relationship with sour mandarin (Citrus sunki Hort.ex Tanaka) and Shatangju mandarin, which are widely cultivated in southern China nowadays, whereas the evolution of mandarins in the northern of China were related to some primitive and old cultivars. In addition, hybrid breeding, long-term artificial selection and domestication led to the subdivision formation and increased the genetic diversity of mandarin. Besides, results of this study showed that GBS has a potential advantage to identify some mandarin accessions with suspicious parents. For example, the phylogenetic tree clearly shows that “Or” tangor has a very close relationship with Kinnow mandarin. 【Conclusion】GBS technology provides an effective and high feasible approach to assistant the taxonomic classification of 240 mandarin accessions, the classification results are in accordance with the conclusion based on morphological method. Meanwhile GBS also is a powerful tool for germplasm identification, and can be applied in the new cultivars identification and intellectual property protection.
GBS; mandarin; phylogenetic evolution; germplasm identification
2016-11-16;接受日期:2017-02-10
国家“十二五”科技支撑计划(2013BAD01B04)、重庆市科委重点项目(cstc2016shms-ztzx80004)
联系方式:王小柯,Tel:18375638987;E-mail:wangxiaoke9191@163.com。通信作者江东,Tel:13983194771;E-mail:jiangdong@cric.cn
猜你喜欢
现代临床医学(2023年1期)2023-03-24 08:30:06
中国医药科学(2022年5期)2022-05-05 23:58:07
中国饲料(2022年5期)2022-04-26 13:42:38
现代园艺(2017年23期)2018-01-18 06:57:52
浙江柑橘(2016年2期)2016-03-11 20:12:41
浙江柑橘(2016年2期)2016-03-11 20:12:40
中国卫生标准管理(2015年3期)2016-01-14 03:41:45
湖南农业科学(2015年5期)2015-02-27 14:33:56
安徽医药(2014年4期)2014-03-20 13:13:40
植物营养与肥料学报(2012年1期)2012-10-26 02:49:46
