
重症肌无力合并多发性肌炎两例临床分析并文献复习
2017-06-01 12:20:40操亚云桂梦翠季苏琼卜碧涛
中国神经免疫学和神经病学杂志 2017年3期
操亚云 桂梦翠 季苏琼 卜碧涛
重症肌无力合并多发性肌炎两例临床分析并文献复习
操亚云 桂梦翠 季苏琼 卜碧涛
目的 观察重症肌无力(MG)合并多发性肌炎(PM)的临床特征及疗效,并探讨其共病机制及治疗方法,提高临床识别度。方法 报道作者医院收治的两例MG合并PM患者的临床资料,结合文献分析其发病特点、实验室检查、疗效及预后等。结果 两例患者临床症状以四肢无力、球肌麻痹、肌肉疼痛为主要表现,疲劳现象不明显,实验室检查可见肌酶升高,偶有心肌受累,肌电图提示肌源性损害;单用溴吡斯的明治疗效果不佳,联合激素和免疫制剂治疗反应良好。结论 MG合并PM的病例少见;MG患者在规范化治疗过程中若出现肌肉疼痛、肌酶谱升高,或疲劳现象消失、眼肌症状不明显等表现时,需行肌电图、肌活检等明确有无合并肌肉疾病的可能;治疗上以免疫治疗为主,急重症者建议使用丙种球蛋白或血浆置换等方法。
重症肌无力;多发性肌炎;临床特点;发病机制
重症肌无力(myasthenia gravis,MG)是一种免疫介导的神经-肌肉接头传递功能障碍的自身免疫性疾病,以B淋巴细胞介导的体液免疫为主,细胞免疫依赖、补体参入等机制在该病中也起着重要作用。多发性肌炎(polymyositis,PM)是一种自身免疫介导的、以侵犯横纹肌为主的全身炎症性疾病,以T淋巴细胞介导的细胞免疫为主,细胞因子等亦在其发病过程中起着重要作用。二者作为自身免疫性疾病,均以肌无力为主要表现,各自有不同特点,两者并发在临床上并不多见。本文报告两例MG合并PM患者的临床特征,并结合文献进行复习,旨在加深临床对其认识,提高临床识别能力。
1 病例报告
病例1,女,19岁,因“眼睑下垂17年,四肢无力1年”于2014-03收入作者医院。患者2岁时出现双眼睑下垂,呈波动性、易疲劳,持续活动后加重,休息后可缓解,有晨轻暮重现象,在作者医院行重复神经电刺激(RNS)检查提示低频递减,新斯的明试验阳性,诊断MG。患者长期口服溴吡斯的明和激素治疗,症状可缓解,14岁时病情完全缓解稳定而停药,后续4年随访中MG症状未复发。1年前患者逐渐出现四肢乏力,表现为上下楼梯困难、梳头费力,无力症状无晨轻暮重现象,不伴眼睑下垂及复视,无吐词不清,无吞咽及呼吸费力。入院前3个月患者肢体无力症状进行性加重,生活自理能力丧失,外院使用小剂量激素后症状稍好转,后转至作者医院就诊。门诊行肌电图检查提示肌源性损害,血生化检查示肌酶谱升高。遂入院进一步诊治。入院查体:双上肢近端肌力Ⅲ级,远端肌力Ⅳ-级,双下肢近端肌力Ⅱ+级,远端肌力Ⅲ+级,双下肢肌肉压痛。入院后行血生化检查显示,肌红蛋白(750.00 ng/mL)、肌酸激酶(4277 U/L)、谷丙转氨酶(51 U/L)、谷草转氨酶(79 U/L)、乳酸脱氢酶(707 U/L)升高。免疫全套检查结果显示,补体C3(0.76 g/L)、补体C4(0.13 g/L)下降。风湿全套检查示抗核抗体胞浆颗粒型1∶320。血、尿、便常规以及肾功能、肿瘤、感染、代谢指标检查均未发现异常。乙酰胆碱受体抗体(AChR-Ab)检测0.1893 nmol/L(阴性)(ELISA法,英国RSR公司试剂盒)。胸部CT检查结果正常。行左侧股四头肌肌肉活检,病理结果提示苏木素-伊红(HE)染色下可见坏死、萎缩、再生肌纤维,坏死肌纤维可见吞噬现象,亦可见明显炎性反应细胞浸润;免疫组化可见CD8/MHC-1分子复合物。排除先天性、代谢性、中毒性、肿瘤性等肌肉疾病,诊断PM。经激素和免疫抑制剂治疗后,患者肢体无力症状好转,肌肉疼痛逐渐减轻,血肌酶亦逐渐下降至正常。后续随诊过程中,患者口服激素及免疫抑制剂,症状控制稳定、肌酶正常、肌痛消失,恢复正常日常活动。
病例2,女,52岁,因“眼睑下垂、四肢无力6年,加重10 d”于2015-07收入作者医院。患者6年无明显诱因出现眼睑下垂、四肢无力,呈波动性、易疲劳,持续活动后加重,休息后可缓解,有晨轻暮重现象。行RNS检查提示低频递减,新斯的明试验阳性,胸部CT检查结果示胸腺瘤,诊断MG、胸腺瘤。2009-09于作者医院胸外科行胸腺瘤切除术,术中发现肺、胸膜转移,行病理检查提示B3型胸腺瘤,行放疗、化疗综合治疗。于2011-06治疗结束,后未复诊。此次入院前因感冒后肌无力症状加重,伴双下肢肌肉酸痛,并伴有吞咽困难、胸闷气短、呼吸费力,口服溴吡斯的明后症状无改善,无力症状无晨轻暮重,无眼睑下垂。入院查体:呼吸急促,平卧不能,双上肢肌力近端Ⅲ+级,远端肌力Ⅳ级,双下肢近端肌力Ⅱ+级,远端肌力Ⅲ级,双下肢肌肉无压痛。入院后行血生化检查结果示,肌红蛋白>1200.00 ng/mL、肌酸激酶(4103 U/L)、谷丙转氨酶(347 U/L)、谷草转氨酶(306 U/L)、乳酸脱氢酶(1304 U/L)升高。肿瘤标志物全套示鳞状细胞癌相关抗原升高(11.4 ng/mL)。AChR-Ab阳性(2.3496 nmol/L)。胸部CT检查未见胸腺瘤复发征象。入院后患者症状进行性加重,考虑MG危象而转入神经科ICU行气管插管、呼吸机辅助呼吸治疗。经干涸疗法、丙种球蛋白、激素、免疫抑制剂等治疗后,患者病情稳定并稍有好转,肌酶谱较前下降,后因经济因素要求转当地医院继续治疗。
患者肌酶谱升高,考虑急性肌肉损伤所致,肌病不能排除,后在作者医院门诊行右侧股四头肌外侧头肌肉活检,病理结果显示,HE染色下可见坏死、萎缩、再生肌纤维,坏死肌纤维可见吞噬现象,可见少许炎性反应细胞浸润;免疫组化可见CD8/MHC-1分子复合物(图1)。同时排除肿瘤、代谢性肌病、药源性肌病等,最终诊断PM。在后续随访过程中,患者长期口服溴吡斯的明、激素、免疫抑制剂,症状基本控制稳定,肌酶降至正常,生活恢复自理。
2 讨论
2.1 临床特点 依据MG诊断和治疗指南[1],本文两例患者临床表现为眼睑下垂、四肢无力、吞咽困难、呼吸费力等,症状呈波动性,易疲劳,持续性活动后加重,休息或使用胆碱酯酶抑制剂后缓解,同时结合RNS、新斯的明试验、MG相关抗体检测等可确诊MG。病例1确诊MG后17年出现四肢无力、肌肉疼痛等症状,病例2确诊MG后6年后出现呼吸吞咽受累、肌肉损伤及酸痛等症状,两者均出现肌肉疼痛、胆碱酯酶抑制剂疗效不佳等非典型MG表现,需完善肌酶检测、肌电图等确定有无合并肌病可能。其中病例2既往有B3型胸腺瘤,肌肉病理提示炎性反应细胞浸润不明显,需排除肿瘤相关的坏死性肌病和免疫介导的坏死型肌病,但患者胸部CT未见肿瘤复发征象,且肌肉免疫组化可见CD8/MHC-1分子复合物,活检中难以发现炎性细胞浸润可能与活检前大剂量使用激素有关[2],综合分析考虑MG合并PM[3]。后上述两例患者均经激素及免疫抑制治疗,病情好转,生活恢复自理,近1年随访病情控制稳定。提示MG患者在规范的治疗过程中,若出现肌肉酸痛、疲劳现象消失、眼肌症状弱化等症状,或病情急速加重时,需考虑合并肌病或其他疾病可能。MG和肌炎均可出现球肌麻痹症状,如吞咽困难和构音障碍等,肌炎患者出现此类症状的比例不足40%[4],MG患者出现此类症状的比例约占发病人数的2/3[5],可见MG患者出现此类症状的比例更高,故PM患者若出现明显的球肌麻痹、眼睑下垂、复视等表现,除常规行头部磁共振排除颅内病变外,也需考虑MG等的可能[6]。
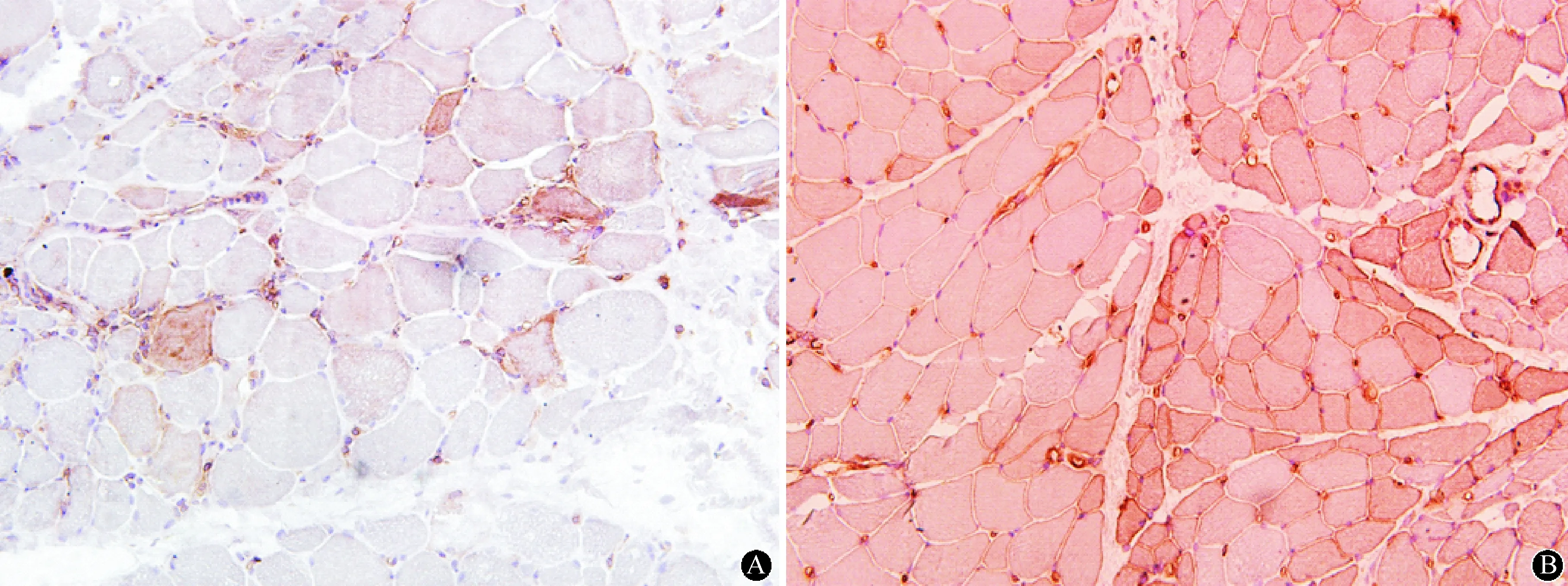
图1 病例2右侧股四头肌外侧头肌肉活检病理学检查可见CD8阳性的T细胞浸润(A,CD8染色×20)以及MHC-1表达阳性的肌膜(B,MHC-1染色×20)
2.2 发病机制 MG合并PM病例少见,国内外报道病例数不足30例。二者共病的机制可能为:(1)细胞因子:MG患者体内的干扰素(IFN)、肿瘤坏死因子(TNF)等细胞因子,可通过促炎反应,放大免疫识别和增强巨噬细胞活性,从而诱导和激发PM发病过程[7];(2)补体:MG患者体内存在AChR-Ab等多种抗体可激活补体,引起肌纤维坏死及炎症细胞浸润,从而导致PM的发生[8];(3)抗体:MG相关抗体众多,抗横纹肌抗体中的连接素抗体(Titin-Ab)和兰尼碱受体抗体(RyR-Ab)被指出与肌肉受累有关[9],二者通过抑制肌浆网钙离子的释放,从而扰乱肌肉的兴奋-收缩耦联机制[9],可能与患者合并肌炎相关[10],但遗憾的是本文两例患者因经济因素未能行Titin-Ab和RyR-Ab检测;(4)胸腺瘤:胸腺作为一种中枢免疫器官,其功能异常(如胸腺瘤)与许多自身免疫性疾病相关[11],尽管目前关于胸腺瘤与肌炎发病机制的研究较少,但文献报道的MG合并PM病例多数伴有胸腺瘤[12],病例2既往有B3型胸腺瘤,尽管此次入院时未见明显胸腺瘤复发征象,但残存胸腺瘤成分中的T淋巴细胞侵袭性浸润不能排除,也可能与胸腺瘤患者更易产生抗骨骼肌成分的抗体有关[13];(5)线粒体:据文献报道线粒体功能异常可能与MG合并PM或皮肌炎有关,如线粒体抗体攻击骨骼肌成分[14],但线粒体抗体往往会引起更广泛的多系统受累。
2.3 疗效及预后 胆碱酯酶抑制剂是治疗MG的首选药物[1],而PM患者一般使用激素与免疫抑制剂[3]。两病共存时,在使用胆碱酯酶抑制剂的基础上推荐小剂量激素起始,逐渐加量至症状改善,建议尽早联合应用免疫抑制剂。由于大剂量激素治疗可能加重MG症状,所以对于以PM首发病的患者,需尽可能地尽早识别合并MG,以避免大剂量激素治疗过程中导致症状恶化。对于急性重症者,丙种球蛋白仍是治疗的首选。文献报道血浆置换治疗疗效肯定[16],但仍有待更多的临床研究证实。MG和PM均属于自身免疫性疾病,治疗原则基本一致,经过积极正规的治疗,一般预后尚可。
综上所述,MG合并PM临床少见,依据其各自的诊断标准及肌电图、肌肉活检结果等不难确诊。二者并发的发病机制尚不明确,考虑与抗体、补体、细胞因子、胸腺瘤等相关。MG合并自身免疫性疾病常见,如自身免疫性甲状腺疾病、类风湿性关节炎,但合并PM少见[5]。MG合并PM往往意味着机体存在更广泛的免疫系统紊乱[17],需尽早识别,尽早启用强有力的免疫治疗,以改善预后。
[1]中华医学会神经病学分会神经免疫学组, 中国免疫学会神经免疫学分会. 中国重症肌无力诊断和治疗指南2015[J].中华神经科杂志,2015,48(11): 934-940.
[2]Lundberg I, Kratz AK, Alexanderson H, et al. Decreased expression of interleukin-1alpha, interleukin-1beta, and cell adhesion molecules in muscle tissue following corticosteroid treatment in patients with polymyositis and dermatomyositis.[J].Arthritis Rheum,2000,43(2): 336-348.
[3]中华医学会神经病学分会, 中华医学会神经病学分会神经肌肉病学组, 中华医学会神经病学分会肌电图及临床神经生理学组. 中国多发性肌炎诊治共识[J].中华神经科杂志,2015,48(11): 946-949.
[4]Marie I, Menard JF, Hatron PY, et al. Intravenous immunoglobulins for steroid-refractory esophageal involvement related to polymyositis and dermatomyositis: A series of 73 patients[J].Arthrit Care Res,2010,62(12): 1748-1755.
[5]Silvestri NJ, Wolfe GI. Myasthenia Gravis[J].Semin Neurol,2012,32(3): 215-226.
[6]Paik JJ, Corse AM, Mammen AL. The co-existence of myasthenia gravis in patients with myositis: A case series[J]. Semin Arthritis Rheu,2014,43(6): 792-796.
[7]Shelton GD, Calcutt NA, Garrett RS, et al. Necrotizing myopathy induced by overexpression of interferon-gamma in transgenic mice[J].Muscle Nerve,1999,22(2): 156, 165.
[8]Gilhus NE, Aarli JA, Matre R. Myasthenia gravis. Antibodies to skeletal muscle cell surface antigens[J].J Neuroimmunol,1983,5(3): 239-249.
[9]Skeie GO, Romi F, Aarli JA, et al. Pathogenesis of myositis and myasthenia associated with titin and ryanodine receptor antibodies.[J].Ann NY Acad Sci,2003,998(998): 343-350.
[10]De BM, Stassen MH. The role of antibodies in myasthenia gravis.[J].J Neurol Sci,2002,202(202): 5-11.
[11]Müller-Hermelink HK, Marx A. Thymoma[J].Curr Opin Oncol,2000,12(5): 426-433.
[12]Behan WMH, Behan PO, Doyle D. Association of myasthenia gravis and polymyositis with neoplasia, infection and autoimmune disorders[J].Acta Neuropathol,1982,57(2-3): 221-229.
[13]Otton SH, Standen GR, Ormerod IE. T cell lymphocytosis associated with polymyositis, myasthenia gravis and thymoma.[J].Clin Lab Haematol,2000,22(5): 307-308.
[14]Shichijo K, Mitsui T, Kunishige M, et al. Involvement of mitochondria in myasthenia gravis complicated with dermatomyositis and rheumatoid arthritis: a case report[J]. Acta Neuropathol,2005,109(5): 539-542.
[15]Seybold ME, Drachman DB. Gradually increasing doses of prednisone in myasthenia gravis. Reducing the hazards of treatment.[J].New Engl J Med,1974,290(2): 81-84.
[16]Ikeda Y,Tanaka M,Mizushima K, et al. A case of eosino philic polymyositis complicated by myasthenia gravis[J]. Muscle Nerve,1998,21(10): 1356-1358.
[17]Aarli JA. Inflammatory myopathy in myasthenia gravis[J]. Curr Opin Neurol,1998,11(3): 233-234.
(本文编辑:时秋宽)
Polymyositis associated with myasthenia gravis: clinical features of two cases and a literature review
CAOYayun,GUIMengcui,JISuqiong,BUBitao*.
*Department of Neurology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan Hubei 430000, China
Corresponding author: BU Bitao, Email: bubitao@tjh.tjmu.edu.cn
Objective To describe the clinical features and therapeutic effects of myasthenia gravis (MG) patients accompanying polymyositis (PM), to discuss the comorbid mechanism and treatment, and to improve the clinical recognition. Methods Through analyzing two cases of hospitalized patients of MG associated with PM in the authors’ hospital and searching the literature of MG associated with PM, we analyzed the characteristics of the onset of the diseases, laboratory examination results, therapeutic effects and prognosis. Results Two patients’ clinical symptoms are mainly limbs weakness, external ophthalmoplegia and muscle pain. The fatigue phenomenon can be unobvious. Laboratory examination shows elevated muscle enzymes associated with occasional myocardial involvement. Electromyography indicates myopathic change. There is a good response only to the treatment of oral prednisolone combined with immunosuppressive agents, but not to pyridostigmine bromide.Conclusions MG combined with PM is rare. When relevant symptoms appeared in the process of normalization treatment for MG patients, such as muscle pain, elevated muscle enzymes, disappearance of fatigue phenomenon, unobvious eye muscle symptoms, electromyography and muscle biopsy should be considered to determine whether muscle disease exists. Treatment with immunotherapy is given in priority when both diseases concur. Intravenous immunoglobulin or plasma exchange treatment should be considered in emergency and crisis.
myasthenia gravis; polymyositis; clinical characteristics;pathogenesis
10.3969/j.issn.1006-2963.2017.03.010
430000 华中科技大学同济医学院附属同济医院神经内科
卜碧涛,Email:bubitao@tjh.tjmu.edu.cn
R746.1
A
1006-2963(2017)03-0193-04
2016-09-18)
猜你喜欢
现代临床医学(2022年2期)2022-04-19 12:48:52
中国民间疗法(2021年17期)2021-11-04 08:39:30
中国临床医学影像杂志(2019年5期)2019-08-27 02:47:48
上海农业学报(2017年3期)2017-04-10 12:39:26
反射疗法与康复医学(2017年7期)2017-01-16 01:11:02
实用中医药杂志(2015年2期)2015-12-01 07:06:18
肿瘤预防与治疗(2015年2期)2015-09-26 07:33:50
中国当代医药(2015年16期)2015-03-01 02:03:13
中国卫生标准管理(2015年15期)2015-01-26 20:32:38
中国药理学通报(2014年2期)2014-05-09 08:22:39
