
高效液相色谱法测定不同厂家克拉霉素颗粒的溶出度
2017-05-18 01:35隋思博宫丽婷
临床医药文献杂志(电子版) 2017年13期
隋思博,宫丽婷
(吉林省药品检验所,吉林 长春 130033)
高效液相色谱法测定不同厂家克拉霉素颗粒的溶出度
隋思博,宫丽婷
(吉林省药品检验所,吉林 长春 130033)
目的采用HPLC法测定不同厂家克拉霉素颗粒的溶出度。方法采用Hibar Purospher STAR C18(250×4.6 mm,5 μm)色谱柱,柱温:45℃,以磷酸盐缓冲液-乙腈(600:400)为流动相,检测波长为210 nm;流速为1.0 ml/min。结果克拉霉素在687.5~5500 μg进样范围内有良好的相应关系,回归方程为y=60.663x+1540.5,r=0.9992。结论HPLC法具有准确度高、重复性好、快速、精密的优点,更加适合克拉霉素颗粒溶出度的测定。
高效液相法;克拉霉素颗粒;溶出度
克拉霉素属于大环内酯类抗生素,主要通过与敏感细菌核蛋白体的50S亚基结合,抑制敏感细菌蛋白合成,发挥抑菌或杀菌作用。适用于治疗幽门螺杆菌感染、呼吸系统感染、非淋菌性泌尿生殖道感染以及皮肤软组织感染等。
2015年版中国药典二部收录了克拉霉素颗粒,溶出度采用的方法是将克拉霉素与75%硫酸反应测定其吸光度,通过试验我们知道,硫酸反应操作复杂且效果不好。本文通过建立高效液相色谱法使试验操作简单,使方法更加准确、高效。
1 仪器与试药
岛津LC-2010 CHT全自动高效液相色谱仪;RC-806溶出试验仪;克拉霉素(中国药品生物制品鉴定所,130558-200902,97.9%);醋酸钠、三乙胺、磷酸二氢钾、冰醋酸、磷酸、乙腈、超纯水、0.45纳米微孔滤膜;克拉霉素颗粒(海南惠普120402,130802;天津红日1212261,1302011,1309241;湖北东信130101;湖北华世通潜龙130813)。
2 方法与结果
2.1 溶出度测定法
以0.1 mol/L醋酸盐缓冲液(pH7.0)(取无水醋酸钠82 g,加水7500 mL,用冰醋酸调节pH值至7.0,加水使成10000 mL)900 mL为溶出介质,转速50转/min,30 min时取样,用溶出介质稀释制成每1mL中含55 μg的溶液。
2.2 色谱条件与系统适应性条件
流动相:磷酸盐缓冲液(取磷酸二氢钾9.11 g,加水溶解并稀释至1000 mL,加三乙胺2 mL,用磷酸调节pH至5.5)-乙腈(600:400),检测波长为210 nm;流速为1.0 ml/ min,柱温:45oC,Hibar Purospher STAR C18 (250×4.6 mm,5μ m)色谱柱;进样量为50 μL。克拉霉素峰的理论板数计算应高于3000,拖尾因子应小于2,克拉霉素峰与相邻前杂质峰的分离度应大于1.5,限度为80%。
2.3 配置对照品
精密称取克拉霉素对照品适量(约相当于主成分22 mg),置100 mL量瓶中,先加少量乙腈振摇使溶解,再加溶出介质稀释至刻度,精密量吸取12.5 mL置50 mL量瓶中,加溶出介质,制成对照品溶液(55 μg/mL)。
2.4 检测限与定量限检验
检测限:精密量取对照品溶液1 mL,置200 mL量瓶中,用溶出介质稀释至刻度,摇匀,作为检测限溶液,精密量取20 μL注入液相色谱,此时信噪比为3:1,克拉霉素四种介质溶液(水、ph1.2缓冲液、ph5.0缓冲液、ph6.8缓冲液)进样量均为5.5 ng。
定量限:精密量取检测限溶液1 mL,置200 mL量瓶中,用溶出介质稀释至刻度,摇匀,作为定量限溶液,精密量取20 μL注入液相色谱,此时信噪比为10:1,硝酸异山梨酯四种介质溶液进样量均为13.75ng。
2.5 耐用性
柱温45℃。①采用Hibar Purospher STAR C18(250×4.6 mm,5 μm)色谱柱测定。②采用Kromasil Eternity (250×4.6 mm,5 μm) PN:EH50704色谱柱测定。③采用Agilent Eclipse XDB-C18(250×4.6 mm,5 μm)PN:990967-902色谱柱测定。采用上述三根不同厂家色谱柱柱进行试验,结果均能很好的分离,可以用于克拉霉素颗粒的测定。
2.6 线性关系与回归方程
精密量取对照品溶液2.5 mL、5 mL、7.5 mL、10 mL、15 mL、20 mL,分别置不同的20 mL量瓶中,用溶出介质稀释至刻度,制成浓度分别为6.875 μg/mL、13.75 μg/mL、20.625 μg/mL、27.5 μg/mL、41.25 μg/mL、55 μg/mL的对照品溶液,各精密吸取100 μL注入液相色谱仪,横坐标为对照品进样量(μg),纵坐标为峰面积,计算回归方程并且绘制标准曲线,可以得出,克拉霉素在687.5~5500 μg进样范围内有良好的相应关系,回归方程为y=60.663x+1540.5,r=0.9992。见表1。
2.7 精密度实验
取对照品溶液,重复6次进样,50 µl/次,结果符合要求。见表2。
2.8 回收率实验
精密称取克拉霉素对照品22.12 mg,置100 mL量瓶中,加醋酸盐缓冲液(pH7.0)稀释至刻度,加乙腈10 mL,振摇使溶解,精密量取12.5 mL,置50 mL量瓶中,用醋酸盐缓冲液(pH7.0)稀释,制成回收对照品溶液(55μg/mL);另精密称取克拉霉素原料(浙江国邦药业有限公司)约11 mg、17.6 mg、22 mg各3份(50%、80%、100%),分别置100 mL量瓶中,加入处方量的辅料,加乙腈10 mL,振摇使溶解,用醋酸盐缓冲液(pH7.0)稀释至刻度,摇匀,滤过,精密量取12.5ml,置50 mL量瓶中,用醋酸盐缓冲液(pH7.0)稀释,制成供试品溶液;吸取回收对照品溶液和供试品溶液各50 μL,注入液相色谱仪,计算回收率。见表3。

表1 标准曲线数据
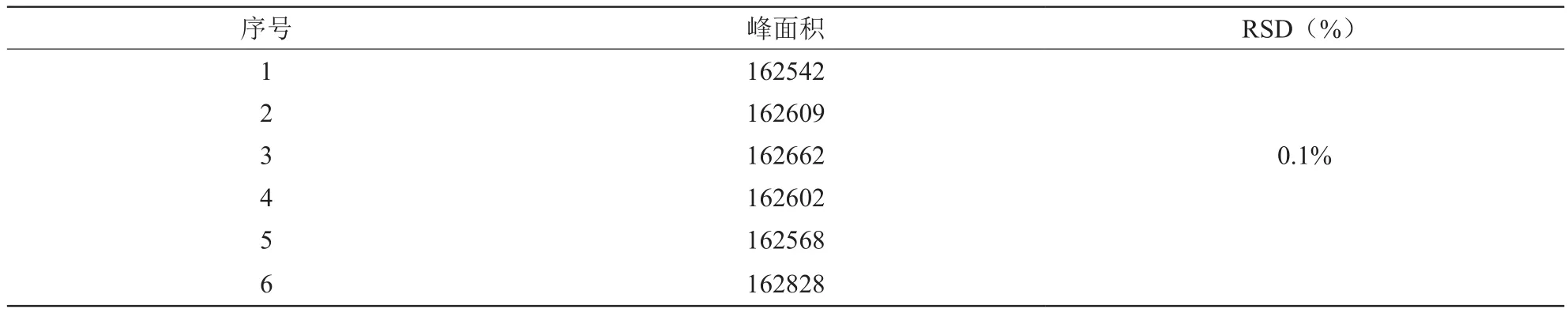
表2 精密度实验结果
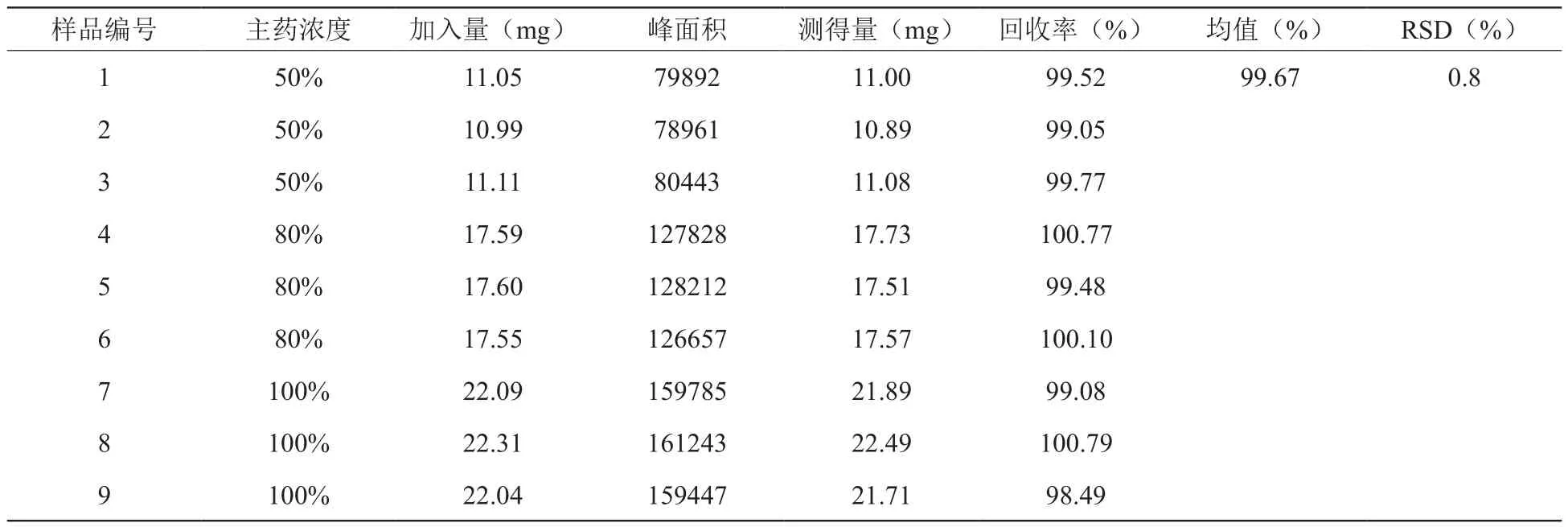
表3 克拉霉素在醋酸盐缓冲液(pH7.0)的回收率结果(n=9)
2.9 溶出度曲线对比
取克拉霉素颗粒(生产厂家为湖北华世通潜龙,批号:130813)照中国药典2015年版溶出度第二法测定,以0.1 mol/L醋酸盐缓冲液(pH7.0)900 mL为溶出介质,转速为50转/min,分别在以下时间取样:5 min、10 min、15 min、20 min、30 min、45 min,滤过。分别量取50μl注入高效液相色谱仪,取克拉霉素对照品溶液(55μg/ml)50μl注入高效液相色谱仪,用峰面积以外标法计算,绘制溶出曲线。同时,按照中国药典2015年版二部克拉霉素颗粒溶出度方法(硫酸显色UV比色法检测),绘制溶出曲线(见图2)。
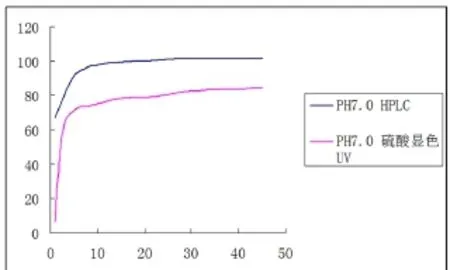
图2 克拉霉素颗粒硫酸显色UV法与PLC测定结果比较
比较两种方法的溶出曲线,可明显的看出HPLC法与UV法存在一定差异。HPLC溶出曲线更加平滑,溶出值更大,更能反应真实的溶出量。
2.10 不同厂家克拉霉素颗粒溶出度测定
取不同厂家的供试品,照中国药典溶出度第二法检验,30min时取样,滤过,用溶出介质稀释制成55μg/ml的供试液,注入高效液相色谱仪,计算溶出度。同时与UV比色法的数据进行对比,结果。可以看出,HPLC法,结果准确、差异更小。见表4。
3 结论与讨论
通过实验与查阅文献[3-5],总结出克拉霉素颗粒溶出度方法为:取本品,照溶出度测定法(附录X C第二法),以0.1 mol/L醋酸盐缓冲液(pH7.0)(取无水醋酸钠82 g,加水7500 mL,用冰醋酸调节pH值至7.0,加水使成10000mL)900 mL为溶出介质,转速为50转/min,依法操作,30 min时,取续滤液适量,用溶出介质定量稀释制成55 μg/mL的溶液,作为供试品溶液;精密称取克拉霉素对照品适量,加少量乙腈溶解,用溶出介质定量稀释制成55 μg/mL的溶液,作为对照品溶液;照含量测定项下的方法试验,精密量取上述两种溶液各50 μL,注入液相色谱仪,记录色谱图,按外标法以峰面积计算溶出量,限度为80%,仅供参考。
通过溶出度曲线的比较、溶出度测定,都可以明确的看出HPLC法优于UC法。克拉霉素与硫酸反应容易受到外界影响,不同的厂家,不同的批号,浓度上的差异,硫酸放置时间,环境的温度,反应是否彻底等等,都会对结果产生影响,无法保证试验的重现性。而HPLC法可以大大节约试验时间,降低操作过程中人为因素的影响,排除硫酸显色中过程不稳定因素的影响,避免了由于硫酸反应不完全带来的干扰, 同时,HPLC法具有准确度高,重复性好的优点。能够使克拉霉素颗粒溶出试验更加准确、科学。
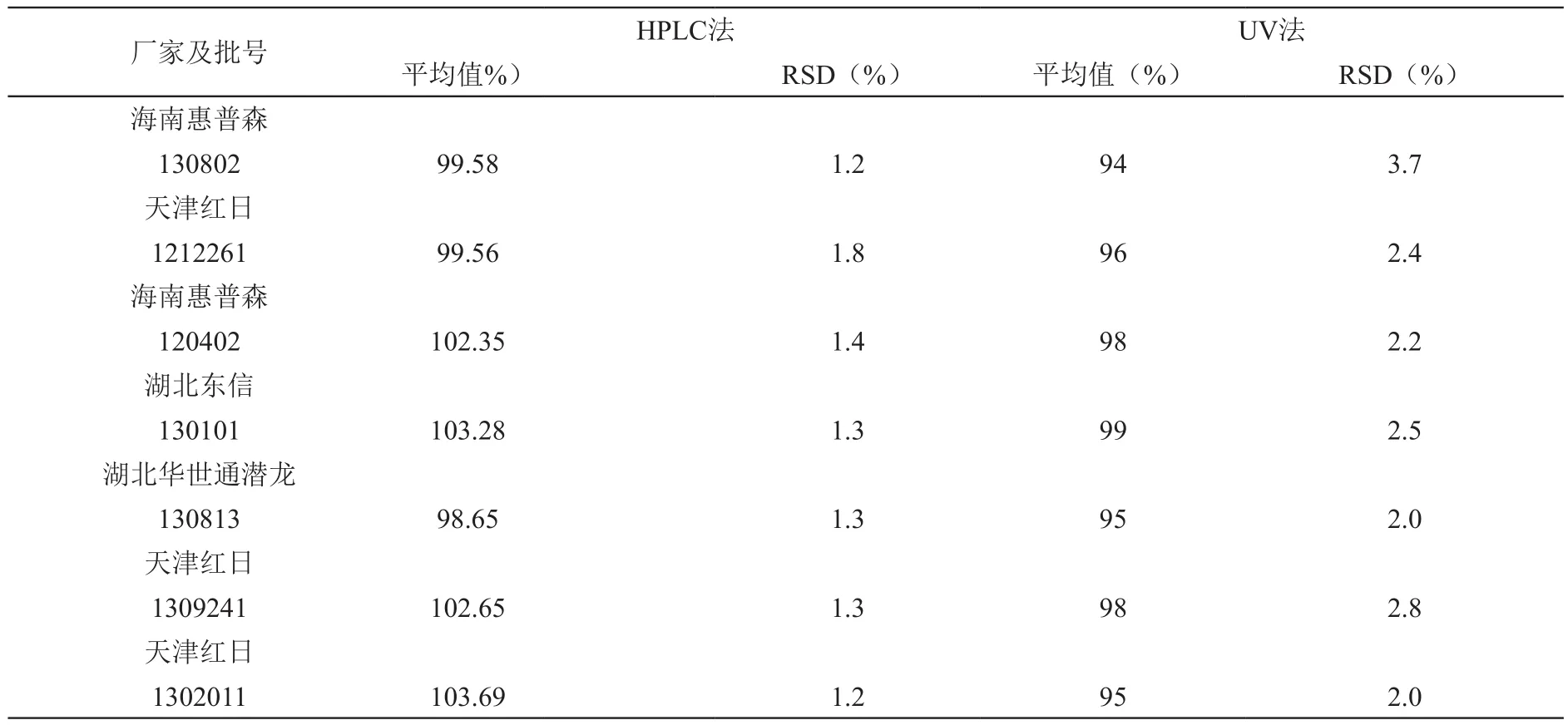
表4 克拉霉素颗粒溶出度测定结果
[1] 吴美芳.高效液相色谱法测定克拉霉素片的溶出度[J].中国药师,2008,12:1525-1526.
[2] 王 巍,曹盛宗,王 勇.高效液相色谱法测定克拉霉素微丸的溶出度[J].海峡药学, 2010,03:48-50.
[3] 汲 静,赵甜爽. 高效液相色谱法测定克拉霉素片的溶出度[J].黑龙江科技信息,2013,22:19.
本文编辑:李 豆
R927.2
B
ISSN.2095-8242.2017.13.2393.03
猜你喜欢
中国药学药品知识仓库(2022年13期)2022-07-03
中国宝玉石(2021年5期)2021-11-18
临床医药文献杂志(电子版)(2020年75期)2021-01-21
大众电视(蓝天下)(2018年8期)2018-10-26
中成药(2018年1期)2018-02-02
中成药(2017年10期)2017-11-16
中成药(2017年10期)2017-11-16
读写算·小学低年级(2017年7期)2017-08-11
特产研究(2016年3期)2016-04-12
天津药学(2013年2期)2013-12-23
