
DAPK1在急性肺损伤小鼠肺组织中的表达及其作用
2017-05-15 03:35刘霞郭小莉雷传江王关嵩王建春
中华肺部疾病杂志(电子版) 2017年2期
刘霞 郭小莉 雷传江 王关嵩 王建春
·论著·
DAPK1在急性肺损伤小鼠肺组织中的表达及其作用
刘霞 郭小莉 雷传江 王关嵩 王建春
目的探讨死亡相关蛋白激酶1(DAPK1)在急性肺损伤小鼠肺组织中的表达及在炎症失控中的作用。 方法将30只雄性C57小鼠按随机数字表法分为5组:正常对照组、LPS诱导急性肺损伤3、6、12、24 h组。应用2 mg/kg DAPK1抑制剂TC-DAPK 6预处理小鼠后,再用10 mg/kg LPS诱导小鼠肺损伤。HE染色光镜观察小鼠肺组织病理改变;免疫组化检测肺组织中DAPK1的表达和分布;应用RT-PCR和Western blot检测肺组织DAPK1和NF-κB p65的表达;ELISA检测血清炎症因子TNF-α、IL-6水平变化;Kaplan-Meier生存分析对各组小鼠存活时间进行分析。结果LPS致伤组肺组织可见明显病理损伤改变且随时间进展加重,而DAPK1蛋白在正常对照组小鼠肺组织仅少量表达,在急性肺损伤小鼠肺组织中表达随时间进展明显增加;与正常对照组相比,LPS组肺组织DAPK1 mRNA蛋白表达水平均明显增高(P<0.05),血浆炎症因子TNF-α、IL-6均明显升高(P<0.05)。抑制小鼠肺DAPK1表达可明显降低LPS诱导的肺组织中DAPK1和NF-κB p65蛋白水平、血清炎症因子TNF-α、IL-6的水平(P<0.05)。此外,抑制DAPK1可延长LPS致急性肺损伤小鼠的生存期(P<0.01)。 结论DAPK1通过调控NF-κB炎症通路参与了LPS诱导的小鼠急性肺损伤,其可作为潜在的治疗靶点。
死亡相关蛋白激酶1; 急性肺损伤; 核因子κB
急性肺损伤(acute lung injury, ALI) /急性呼吸窘迫综合征(acute respiratory distress syndrome, ARDS) 是临床常见的由严重感染、创伤等多种因素诱发的以难治性低氧血症为特征的危重症[1-3],目前认为多种炎症细胞参与引起的炎症失控导致肺实质弥漫性损伤是ALI /ARDS 的主要病理生理基础[4-5]。
死亡相关蛋白激酶1(death associated protein kinase 1, DAPK1)是一种钙调蛋白(CaM)调节的丝氨酸/ 苏氨酸蛋白激酶,参与机体多种病理生理过程[6]。既往研究表明DAPK1是凋亡的正性调节因子之一,广泛参与多种途径诱导的细胞凋亡[7-8]。近来不断有研究发现其除具有调节细胞凋亡、自噬外,在一系列炎症调节中也具有重要作用[9]。Yoo等[10]研究发现,在卵巢癌细胞株OVCAR3,DAPK1抑制INF-γ、TNF-α诱导的NF-κB激活,可抑制环氧合酶-2和细胞间黏附分子-1(ICAM-1)的产生。为此,本研究采用LPS复制肺损伤小鼠模型,观察肺组织DAPK1的表达及炎症介质的释放情况,并应用DAPK1的特异性抑制剂TC-DAPK 6预处理小鼠后,再观察LPS诱导小鼠的肺损伤严重程度、炎症因子的水平以及小鼠的生存时间,进而探讨DAPK1在LPS诱导的ALI/ARDS中的作用及其机制。
材料与方法
一、试剂与材料
1. 实验动物: 30只雄性6~8周龄C57BL/6小鼠(体质量22±2 g)购自第三军医大学实验动物中心。
2. 主要试剂: HE染色病理切片由新桥医院中心实验室完成,免疫组化试剂购自武汉博士德生物技术有限公司; ELISA 试剂盒购自上海沪尚科技有限公司; 蛋白提取试剂盒、蛋白酶抑制剂、ECL 超敏化学发光显影液均购自美国Thermo 公司;LPS 购自Sigma 公司;SDS-PAGE凝胶试剂盒购自碧云天公司;DAPK1和NF-κB p65兔抗鼠多克隆抗体购自美国Abcam公司;GAPDH 兔多克隆一抗、二抗辣根过氧化物酶标记山羊抗兔购自北京中杉金桥生物有限公司;RT-PCR逆转录试剂盒购自TaKaRa公司;DAPK1抑制剂购自美国MedChemExpresss公司。
二、研究方法
1. 实验动物ALI模型构建: 30只雄性6~8周龄C57BL/6小鼠按随机数字表法分为5组:正常对照组、LPS致肺损伤3、6、12、24 h组。LPS处理组LPS(10 mg/kg)腹腔注射以建立急性肺损伤模型,正常对照组不处理。处理完毕后将动物放回笼内自由活动,不禁食水。小鼠在LPS等其他因素干预后,应用干冰将其窒息死亡,并取相应的标本。
2. 肺组织病理学检查: 取小鼠右肺组织于10%中性甲醛固定后,经脱水、石蜡包埋、切片,HE 染色,最后封固,常规光镜下观察肺组织形态学改变。
3. 免疫组化检测小鼠肺组织DAPK1表达情况: 取小鼠肺组织右肺叶标本行免疫组化S-P 法染色,采用柠檬酸盐缓冲液(0.01 mol/L)高温高压抗原修复,常规透膜,5% BSA 室温封闭1 h,滴加一抗(1︰250) 4 ℃过夜。HRP 标记的二抗(1︰1 000) 37 ℃孵育1 h,DAB显色,自来水冲洗终止,苏木精复染10 min 后返蓝,烤干组织切片,中性树胶封片,镜下观察。
4. Real-time PCR: 使用TRIzol 法提取小鼠肺组织总RNA,微量核酸蛋白检测仪测定RNA浓度。将mRNA 反转录为cDNA,RT-PCR检测各样本DAPK1表达,反应条件: 95 ℃ 30 s,95 ℃ 5 s,56 ℃ 30 s,72 ℃ 30 s,总共38 个循环。以β-actin为内参,每组设3个复孔,重复3次,计算平均值。引物由金斯瑞(南京)生物科技有限公司设计合成。引物序列: DAPK1 上游引物5′-ATGACTGTGTTCAGGCAGG AA-3′,下游引物5′-CCGGTACTTTTCTCACGACATTT-3′,扩增产物为107 bp。
5. ELISA 检测血浆TNF-α、IL-6 水平: 在相应时点眼球取血,4 ℃低温离心(以离心半径8 cm,3 000 r/min,离心10 min),分离出血浆,按照ELISA 试剂盒说明书检测各标本血浆TNF-α、IL-6 蛋白表达水平。每样本和标准品均设双复孔,获取酶标仪450 nm 处光密度(OD)值,并根据OD值绘制标准曲线,计算各样本TNF-α、IL-6 含量,取平均值为统计数据。
6. DAPK1抑制剂预处理小鼠以及Western blot 检测DAPK1、NF-κB p65在肺组织的蛋白表达: 因DAPK1在急性肺损伤小鼠肺组织中表达明显增加,且24 h达到最高,故选择DAPK1特异性抑制剂抑制其表达。DAPK1 选择性抑制剂TC-DAPK 6(2 mg/kg)腹腔注射注入小鼠体内,1 h后再接受LPS(10 mg/kg) 腹腔注射建立肺损伤模型,24 h后检测DAPK1蛋白表达、促炎基因表达及炎症因子表达水平。按蛋白提取试剂盒说明书提取肺组织总蛋白,浓度经微量核酸蛋白检测仪测定。各标本取等量蛋白样品进行8% SDS-PAGE 电泳2 h (恒压80 V) ,转膜(恒流300 mA,70~160 min),5%BSA封闭1 h,分别加入DAPK1和NF-κB p65一抗(1︰1 000) 、GAPDH 一抗(1︰3 000) ,4 ℃过夜。TBST洗膜后加入HRP 标记的二抗(羊抗IgG,1︰5 000)37 ℃孵育1 h ,TBST洗膜后加入ECL化学发光法显色成像。用Quantity One 软件测定各条带积分光密度值,以GAPDH条带校正。
三、统计学方法
结 果
一、小鼠肺组织病理学改变和血清炎症因子TNF-α、IL-6的水平
光镜下观察肺组织HE染色可见:正常对照组小鼠肺组织细胞结构清晰,肺泡壁薄而光滑,边缘完整,肺泡腔及肺间质内无炎症细胞浸润渗出。LPS致肺损伤3、6、12、24 h 组小鼠肺组织均可见肺组织毛细血管充血水肿,肺泡间隔增厚,部分肺泡塌陷,肺间质炎症细胞浸润,部分肺泡腔内可见液体渗出,肺组织结构破坏程度随时间增加而加重(图1A)。检测LPS致肺损伤组小鼠各个时间点的血清炎症因子水平,发现在对照组和LPS处理小鼠血清TNF-α和IL-6水平在3 h时分别为65.59±2.24 ng/L和59.36±2.26 ng/L,6h时分别为174.95±5.50 ng/L和111.39±5.00 ng/L,12 h时分别为376.20±4.88 ng/L和204.12±4.86 ng/L,24 h时分别为379.28±2.69 ng/L和206.09±3.23 ng/L。LPS损伤后,各个时间点的血清TNF-α和IL-6水平均显著高于对照组(P<0.05),并且随着LPS作用时间的延长而升高,但是LPS处理12 h和24 h时相点血清TNF-α、IL-6水平均无显著差异(图1B、C)(P>0.05)。
二、LPS致小鼠ALI后DAPK1肺组织的表达
免疫组化示在正常肺组织中DAPK1少量表达于肺组织,而在LPS处理后,随着肺损伤的逐渐加重,肺组织中表达DAPK1的细胞数量较对照组明显增多(图2A)。Western blot 检测结果显示(图2B),LPS干预小鼠后各个时相点肺组织DAPK1的蛋白表达高于对照组(P<0.05),在24 h时相点表达最高。qPCR检测(图2C)同样发现LPS干预小鼠后各个时相点肺组织DAPK1 mRNA的表达高于对照组(P<0.05),并且在24 h时表达最高。同时,LPS干预小鼠6 h后肺组织中DAPK1 mRNA表达也明显低于3 h、12 h和24 h时的表达(P<0.05)。
三、DAPK1抑制后肺损伤小鼠肺组织中DAPK1、NF-κB p65的表达
本研究使用DAPK1的选择性抑制剂TC-DAPK 6预处理小鼠后,再用10 mg/kg LPS腹腔注射24 h后取标本,病理结果显示(图3A),TC-DAPK 6预处理小鼠后,可明显降低LPS诱导小鼠肺损伤。qPCR检测结果示(图3B),LPS可显著增加DAPK1 mRNA表达(P<0.05),而TC-DAPK 6可显著逆转LPS诱导的DAPK1 mRNA的升高(P<0.05)。Western blot 检测结果显示(图3C),在LPS诱导组DAPK1与NF-κB p65蛋白较对照组呈高表达(P<0.05),而TC-DAPK 6预处理后再用LPS处理,DAPK1与NF-κB p65蛋白的表达显著低于LPS处理组(P<0.05)。
四、DAPK1抑制后肺损伤小鼠血清炎症因子TNF-α、IL-6的变化及生存期分析
本研究使用TC-DAPK 6抑制DAPK1及其NF-κB p65的表达后,用ELISA检测血清TNF-α和IL-6的变化,control组、LPS组和TC-DPAK 6组的血清TNF-α和IL-6水平分别为63.33±2.53 ng/ml和59.86±3.14 ng/ml、378.06±7.59 ng/ml和207.07±6.14 ng/ml、225.68±7.45 ng/ml和146.07±3.34 ng/ml。TC-DPAK 6组和LPS组的血清TNF-α和IL-6水平显著高于Control组(P<0.05),而TC-DPAK 6组的血清TNF-α和IL-6水平明显低于LPS组,P<0.05(图4A、B)。通过Kaplan-Meier生存分析发现,TC-DPAK 6组小鼠的生存期显著长于LPS组,P<0.01(图4C)。
讨 论
ARDS 是临床常见的危重症,其发病机制错综复杂。目前认为多种炎症细胞(如多核白细胞、肺泡巨噬细胞)及肺泡上皮细胞、肺血管内皮细胞等均参与ARDS的发生、进展。其主要机制是通过炎症细胞可释放大量炎症介质,导致肺泡上皮细胞发生结构和功能改变,引起肺不张,通气/血流比例失调;而肺血管内皮细胞受损后,导致血管通透性增加发生肺水肿,引起ARDS 的主要病理改变,继而引发全身炎症反应综合征(systemic inflammatory response syndrome, SIRS),失控的SIRS进一步发展为多器官功能障碍综合征(multiple organ dysfunction syndrome, MODS),ARDS则是MODS的一个重要组成部份。因此,目前认为急性肺损伤ALI/ARDS的本质是一种炎症,其发病与炎症的失控密切相关[4, 11]。尽管近年来人们对 ARDS 研究不断深入,诊疗技术有明显提升,但其病死率仍然居高不下,重度ARDS患者病死率仍高达40%[12]。

图1 LPS致大鼠急性肺损伤;注:A.HE染色观察LPS处理小鼠不同时间点肺组织的损伤(×20);①对照组;② LPS处理3 h;③LPS处理6 h;④LPS处理12 h;⑤ LPS处理24 h;B. ELISA检测血清TNF-α、IL-6水平;*,与control组比较,P<0.05; ^,与LPS处理3 h组比较,P<0.05; #,与LPS处理6 h组比较,P<0.05

图2 LPS致小鼠急性肺损伤各个时相点DAPK1的表达及定位。A.免疫组化染色观察LPS处理小鼠不同时间点对肺组织中DAPK蛋白的定位和表达(×20);注:①对照组;②LPS处理3 h;③LPS处理6 h;④ LPS处理12 h;⑤ LPS处理24 h;B. Western blot检测LPS致小鼠急性肺损伤各个时相点DAPK蛋白的表达情况;C. qPCR检测LPS致小鼠急性肺损伤各个时相点DAPK mRNA的表达情况;*与control组比较P<0.05; ^与LPS处理3 h组比较,P<0.05;#与LPS处理6 h组比较,P<0.05;&与LPS处理12 h组比较,P<0.05
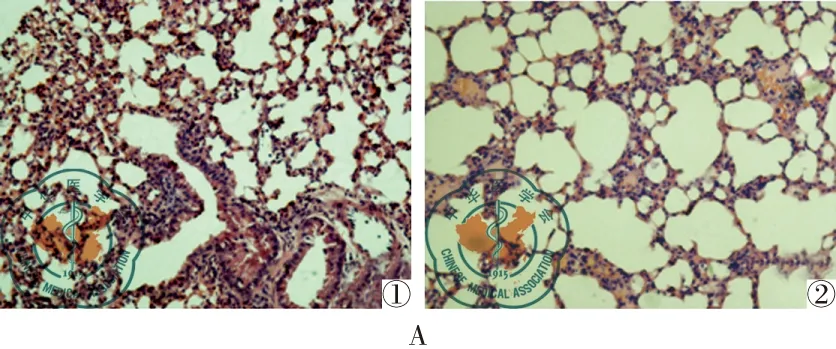
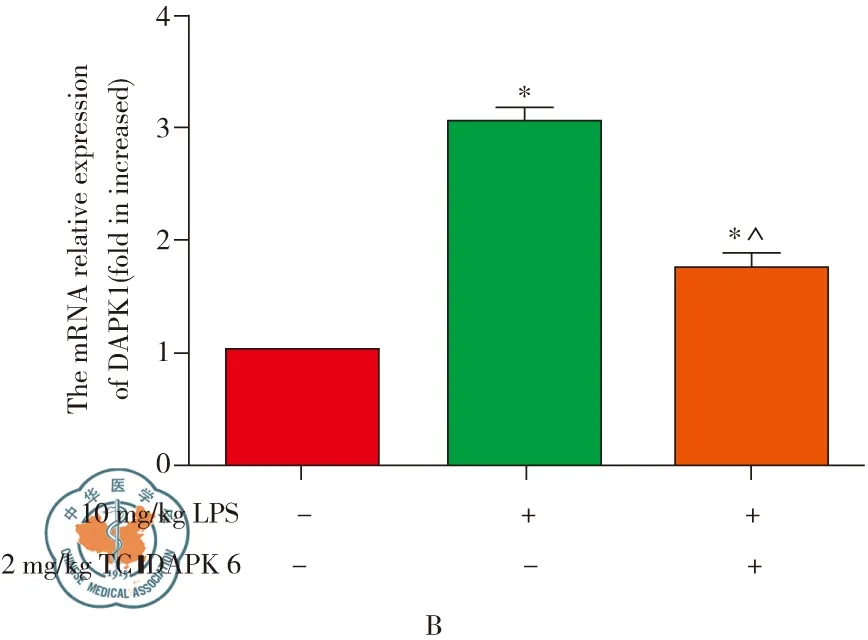
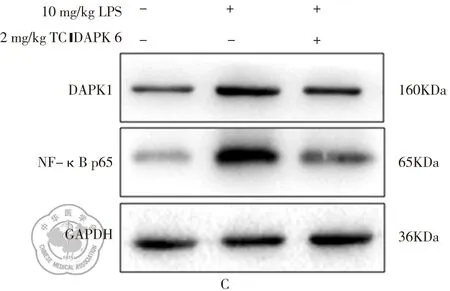
图3 TC-DAPK 6抑制LPS诱导的肺损伤,下调DAPK1及NF-κB p65的表达;注:A: HE染色观察TC-DAPK 6预处理后,LPS诱导小鼠急性肺损伤(×20). 1: 单独LPS组;2:TC-DAPK 6+LPS组;B:qPCR检测DAPK1的表达水平。*与control组比较P<0.05; ^与LPS组比较P<0.05;C:Western blot检测DAPK1和NF-κB p65蛋白的表达情况
DAPK1作为凋亡正性调节因子,广泛参与TGF-β、IFN-γ、TNF-α和Fas等多种途径诱导的细胞凋亡[7-8]。此外,近年来研究发现DAPK1在一系列炎症调节中也具有重要作用。在肠上皮细胞,DAPK1抑制TNF-α诱导的STAT3激活和IL-6产生;DAPK1在溃疡性结肠炎相关慢性炎症中起保护作用;在TNF-α诱发的血管炎也发现,DAPK通过抑制NF-κB 活性发挥抗炎作用;在DAPK敲除鼠模型,给予小剂量LPS经鼻吸入刺激后,肺泡灌洗液中KC、IL-6分泌增加,肺上皮细胞P65核异位增加[13-16]。然而,DAPK1在IL-1β的产生中起重要作用;抑制DAPK1表达可显著减少巨噬细胞诱导的IL-8分泌[17-18]。因此,DAPK1在炎症调节中既有正向调节作用,也有负向调节作用,而这种参与炎症调节的不同作用可能与DAPK1影响NF-κB活性及细胞种类相关[19]。在本实验中,LPS诱导的小鼠肺损伤模型中肺组织DAPK1蛋白和mRNA表达均明显增高,且随时间延长有明显上升趋势,与此同时,血浆炎症因子TNF-α、IL-6也呈现出逐渐升高的趋势。因此,我们推测DAPK1参与了LPS致小鼠急性肺损伤的进程,并且DAPK1的表达可能与炎症因子水平有相关。
既往研究表明,NF-κB在急性肺损伤时表达增多,是调控炎症介质表达的关键转录因子,在细胞受到刺激后进入细胞核与靶基因DNA 接触调控IL-1β、TNF-α、IL-6、IL-1β等炎症介质的表达合成[20]。本研究中同样发现LPS诱导小鼠肺损伤后,NF-κB显著升高。我们采用DAPK1特异性抑制剂抑制小鼠肺DAPK1表达后, 发现NF-κB p65蛋白水平及血清炎症因子TNF-α和IL-6的表达水平均明显下降。提示DAPK1可通过NF-κB调控炎症因子的产生和释放,但具体机制尚不清楚。

图4 TC-DAPK 6下调LPS诱导的小鼠血清炎症因子血清IL-6、TNF-α水平并延长小鼠生存期;注:A和B,ELISA检测血清TNF-α、IL-6水平。*与control组比较,P<0.05; ^与LPS处理组比较,P<0.05;C:Kaplan-Meier生存分析小鼠生存时间。*与LPS组比较,P<0.05
综上所述,本研究结果表明,急性肺损伤小鼠肺组织DAPK1表达明显上升,血浆炎症因子TNF-α、IL-6大量释放,通过抑制DAPK1表达后NF-κB p65蛋白水平及血清炎症因子TNF-α和IL-6的表达水平均明显下降,提示DAPK1参与急性肺损伤的发生发展,加重肺部炎症反应,其机制可能与增强NF-κB 的转录活性相关,但其具体作用及机制还需进一步研究。结合既往研究结果,表明DAPK1在LPS诱导急性ALI/ARDS的发生、发展中发挥着关键作用,其可作为ALI/ARDS治疗的新靶点。
1 金发光. 急性肺损伤的诊治研究现状及进展[J/CD]. 中华肺部疾病杂志(电子版), 2013, 6(1): 1-3.
2 马李杰, 李王平, 金发光. 急性肺损伤/急性呼吸窘迫综合征发病机制的研究进展[J/CD]. 中华肺部疾病杂志(电子版), 2013, 6(1): 65-68.
3 宋旸, 蒋昊翔, 张永红, 等. 急性呼吸窘迫综合征药物研究进展[J/CD]. 中华肺部疾病杂志(电子版), 2015, 8(6): 769-772.
4 Matthay MA, Ware LB, Zimmerman GA. The acute respiratory distress syndrome[J]. J Clin Invest, 2012, 122(8): 2731-2740.
5 Cross LJ, Matthay MA. Biomarkers in acute lung injury: insights into the pathogenesis of acute lung injury[J]. Crit Care Clin, 2011, 27(2): 355-377.
6 Lin Y, Hupp TR, Stevens C. Death-associated protein kinase (DAPK) and signal transduction: additional roles beyond cell death[J]. FEBS J, 2010, 277(1): 48-57.
7 Jang CW, Chen CH, Chen CC, et al. TGF-beta induces apoptosis through Smad-mediated expression of DAP-kinase[J]. Nat Cell Biol, 2002, 4(1): 51-58.
8 Cohen O, Inbal B, Kissil JL, et al. DAP-kinase participates in TNF-alpha- and Fas-induced apoptosis and its function requires the death domain[J]. J Cell Biol, 1999, 146(1): 141-148.
9 Lai M Z, Chen RH. Regulation of inflammation by DAPK[J]. Apoptosis, 2014, 19(2): 357-363.
10 Yoo HJ, Byun HJ, Kim BR, et al. DAPk1 inhibits NF-kappaB activation through TNF-alpha and INF-gamma-induced apoptosis[J]. Cell Signal, 2012, 24(7): 1471-1477.
11 Baue AE. MOF, MODS, and SIRS: what is in a name or an acronym? [J]. Shock, 2006, 26(5): 438-449.
12 Villar J, Sulemanji D, Kacmarek RM. The acute respiratory distress syndrome: incidence and mortality, has it changed?[J]. Curr Opin Crit Care, 2014, 20(1): 3-9.
13 Chakilam S, Gandesiri M, Rau TT, et al. Death-associated protein kinase controls STAT3 activity in intestinal epithelial cells[J]. Am J Pathol, 2013,182(3): 1005-1020.
14 Bajbouj K, Poehlmann A, Kuester D, et al. Identification of phosphorylated p38 as a novel DAPK-interacting partner during TNFalpha-induced apoptosis in colorectal tumor cells[J]. Am J Pathol, 2009, 175(2): 557-570.
15 Usui T, Okada M, Hara Y, et al. Death-associated protein kinase 3 mediates vascular inflammation and development of hypertension in spontaneously hypertensive rats[J]. Hypertension, 2012, 60(4): 1031-1039.
16 Nakav S, Cohen S, Feigelson SW, et al. Tumor suppressor death-associated protein kinase attenuates inflammatory responses in the lung[J]. Am J Respir Cell Mol Biol, 2012, 46(3): 313-322.
17 Chuang YT, Lin YC, Lin KH, et al. Tumor suppressor death-associated protein kinase is required for full IL-1beta production[J]. Blood, 2011, 117(3): 960-970.
18 Turner-Brannen E, Choi KY, Arsenault R, et al. Inflammatory cytokines IL-32 and IL-17 have common signaling intermediates despite differential dependence on TNF-receptor 1[J]. J Immunol, 2011, 186(12): 7127-7135.
19 Chuang YT, Fang LW, Lin-Feng MH, et al. The tumor suppressor death-associated protein kinase targets to TCR-stimulated NF-kappa B activation[J]. J Immunol, 2008, 180(5): 3238-3249.
20 Ali F, Ismail A, Kersten S. Molecular mechanisms underlying the potential antiobesity-related diseases effect of cocoa polyphenols[J]. Mol Nutr Food Res, 2014, 58(1): 33-48.
(本文编辑:王亚南)
刘霞,郭小莉,雷传江,等. DAPK1在急性肺损伤小鼠肺组织中的表达及其作用[J/CD]. 中华肺部疾病杂志(电子版), 2017, 10(2): 151-156.
Expression and function of DAPK1 in lung tissues of mouse with acute lung injury
LiuXia,GuoXiaoli,LeiChuanjiang,WangGuansong,WangJianchun.
DepartmentofRespiratoryDisease,XinqiaoHospital,ThirdMilitaryMedicalUniversity,Chongqing400037,China
WangJianchun,Email:wjc5577@126.com
Objective To investigate the expression of death associated protein kinase 1(DAPK1) in lung tissues of mouths with LPS induced acute lung injury(ALI) and its function in ALI. Methods Thirty C57 male mice were randomly divided into 5 groups: control group, LPS induced acute lung injury group for 3 h, 6 h, 12 h and 24 h group. Histological changes of the lungs were observed by HE staining; The expression and distribution of DAPK1 in the lung tissues was detected by western blot, qPCR and Immunohistochemistry. The serum levels of TNF-α and IL-6 were detected by ELISA. Six mice were pretreated with 2 mg/kg TC-DAPK 6, a selectivity DAPK1 inhibitor, and then 10 mg/kg LPS was used to induce mice acute lung injury by intraperitoneal injection. The expression of DAPK1 and NF-κB p65 was evaluated using western blot and qPCR, and ELISA was used to analyze the level of serum TNF-α and IL-6. Kaplan-Meier analysis was evaluated the lifespan of these mice. Results The HE stain showed that pathological of lung tissue damage became severe along with time. Immunohistochemistry demonstrated that. The area of DAPK1 protein expression was more large with time increased in LPS group, compared to control group (P<0.05).The level of serum TNF-α and IL-6 was significantly higher than control group (P<0.05). The expression of DAPK1 and NF-κB p65 and serum TNF-α and IL-6 level was significantly decreased after pretreated with TC-DAPK 6 and stimulated with LPS, compared to LPS alone group (P<0.05). Moreover, Kaplan-Meier analysis showed the lifespan of mice by pretreated with TC-DAPK 6 was longer than LPS alone group (P<0.01). Conclusion DAPK1 involved in LPS induced mice acute lung injury by modulation NF-κB p65 and the production and released of inflammation mediator. It might be a potential target for ALI/ARDS treatment.
Death associated protein kinase 1; Acute lung injury; Nuclear factor-κB
10.3877/cma.j.issn.1674-6902.2017.02.008
国家自然基金面上资助项目(81170066)
400037 重庆,第三军医大学新桥医院呼吸内科
王建春, Email: wjc5577@126.com
R563
A
2017-01-11)
猜你喜欢
九江学院学报(自然科学版)(2022年2期)2022-07-02
新世纪智能(数学备考)(2021年10期)2021-12-21
昆明医科大学学报(2021年10期)2021-12-02
昆明医科大学学报(2021年10期)2021-12-02
中老年保健(2021年5期)2021-08-24
天津医科大学学报(2021年4期)2021-08-21
感染、炎症、修复(2021年1期)2021-07-28
感染、炎症、修复(2021年1期)2021-07-28
科学大众(2021年6期)2021-07-20
新世纪智能(数学备考)(2020年10期)2021-01-04
