
漆酶催化邻苯二酚开环的自由基反应机制
2017-05-10 17:42:42陈明王林谭天罗学才郑在尹若春苏吉虎杜江峰
物理化学学报 2017年3期
陈明 王林 谭天 罗学才 郑在 尹若春 苏吉虎,* 杜江峰
(1中国科学技术大学近代物理系,中国科学院微观磁共振重点实验室,合肥230026;2安徽大学生命科学学院生物制造创意中心,合肥230601)
漆酶催化邻苯二酚开环的自由基反应机制
陈明1王林2谭天1罗学才2郑在2尹若春2苏吉虎1,*杜江峰1
(1中国科学技术大学近代物理系,中国科学院微观磁共振重点实验室,合肥230026;2安徽大学生命科学学院生物制造创意中心,合肥230601)
酶催化反应是绿色化学领域中的研究热点之一。在常用的酶中,漆酶是一种多铜氧化酶。在此,漆酶被用来研究邻苯二酚的氧化和芳香环开环的自由基过程。实验表明,邻苯二酚的初始氧化产物是一个半醌自由基,再发生分子间加成反应而生成二聚体和三聚体(通过质谱鉴定),和分子内加成而生成可被5,5-二甲基-1-氧化吡咯啉(DMPO)所捕获的含有支链烷基自由基的呋喃衍生物。后者经核磁共振氢谱(1H-NMR)加以分析。同时,通过17O同位素标记追踪方法,我们发现水直接参与该分子内加成反应,并释放出17O标记的羟基自由基(·17OH)。此外,除了3-甲基邻苯二酚和4-甲基邻苯二酚两种类似物,此自由基过程在间苯二酚、对苯二酚、萘二酚、3-硝基邻苯二酚和4-硝基邻苯二酚等底物中均未发生。据此,我们对C4-C5位点的选择性活化与开环的机理展开分析和讨论,并将该机理与邻苯二酚双加氧酶导致的邻苯二酚双氧中间位置和相邻位置的开环过程相比较。这些结果将有益于漆酶的改造和仿生。
漆酶;自由基氧化;芳香环开环;电子顺磁共振波谱;自旋捕获技术
1 Introduc tion
Laccase(benzenediol:oxygen oxidoreductase,EC1.10.3.2), belongs to a fam ily ofmulticopper oxidases found in bacteria, fungi,green plants,and insects1-4.Theactive centerof laccase is composed of four inorganic copper ions that are assigned respectively to T1and T2/T3sites(Fig.1).The T1site consistsof one redox-active Cu2+ionwhich is reduced to Cu1+afteraccepting an electron from the substrate donor,and then is turned back to thermal-stable Cu2+after delivering the electron to downstream acceptor;The T2/T3sites contain 3 Cu clusters that reduceO2to waterafteraccepting fourelectrons from T1in steps1-3,5.The redox changes of T1and T2/T3Cu ions coupled w ith electron transfer within laccasehasbeen elucidated1-3,5,6.
Because of the high redox potential,laccase-catalyzed transformations have recently become an active research field in chem istry7-10and biology11-14.For examp le,an ever-increasing focuson laccasemediated reaction has been shown in oxidation, polymerization and adduction reactions1,2,15-17.These processesare always involved with the cleavage and formation of chemical bonds and the electron transfer.Unfortunately,the substrate-dependentmechanism of substrate transformation in T1site isseldom studied.This is thus unclearw hen com pared to the well-established electron transfer pathway from T1to O2via T2/T3sites1-3,5. As for the potential application and protein designment,the mechanism must be elucidated experimentally.Moreover,the product/intermediates released from Cu-T1aremore important than theO2-to-water transformation at T2/T3sites.This question wasaddressed herein with catecholand analogsas laccase substrate.
Among laccase′s substrates,phenolsareof importancebecause of their uses as the building blocks in industry,agriculture, medicine,synthesis and polymer chem istry18.Catechol(o-dihydroxybenzene),a typicalderivativeof phenol,plays a key role in physiologicalactivities18-21.A number of substrate analogs,m-and p-dihydroxybenzene(resorcinoland hydroquinone),3-or4-methyl catechol,3-or4-nitroso catechol,and 2,3-dihydroxynaphthalene, werealso used to study themechanism clearly.
2 Materia ls and m ethods
Laccase(EC1.10.3.2,>10U·mg-1)ispurchased from Sigma-A ldrich.Catechol(99%)and analogs in Scheme 1 are purchased from J&K Scientific.Trapping reactant,5,5-dimethyl-1-pyrroline-N-oxide(DMPO)(>99.0%),is from Dojindo International.The reaction solution with double stilledwater contains laccase and catecholor itsanalogueswith the finalpH~6.3.After5-or15-min reaction,200μL ofm ixture w as sam pled for the follow ing experiments.Analogs(Scheme 1),aswell as m-and p-dihydroxybenzene(resorcinoland hydroquinone),3-or 4-methyl catechol (98%),3-or 4-nitroso catechol(98%),and 2,3-dihydroxynaphthalene(98%),were used as controls.Ethyl acetate was high performance liquid chromatography(HPLC)grade and purchased from Tianjin Guangfu fine chem ical research institute.
After 5-or 15-min procedure reactionsat room temperature, 200μL of solution contains laccase(0.5mg·m L-1)and catechol or its analogues(0.5mg·mL-1)was sampled,and then stored quickly in liquid nitrogen until electron paramagnetic resonance (EPR)measurement.The freeze-trapping EPR spectrawere recordedwith a Bruker X-band EMX EPR spectrometerat20K.
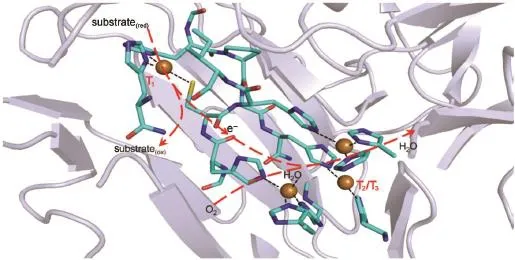
Fig.1 Corestructure and electron transfer in blue laccase from Lentinus tigrinus(PDB code2QT6)
For spin-trapping,200μL of reactionm ixture has laccase(0.25 mg·mL-1),catecholor phenolderivative(0.25mg·mL-1),DMPO(100mmol·L-1).The reactionwaspreceded at room temperature about3-4m in,then trapping reagentDMPO was added sw iftly to trap the radicals for EPRmeasurements(Scheme 2)at room temperature.
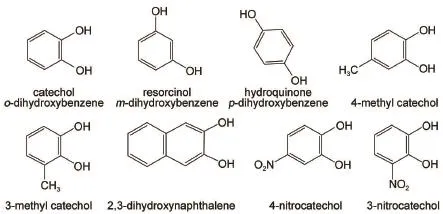
Schem e 1 Structures of catecholand analogs
HPLC-MS(massspectrometry)wasperformedwith theWaters Xevo G2 QTof.The catalytic reaction containing 1mg·m L-1laccase and the corresponding substratesw ere preceded about 5 min at room temperature.The sampledm ixturewas filtered first through a0.45-μm filtermembranebefore injection.The HPLCMSsettingswere:mobile phase,acetonitrile/water(20%/80%); flow velocity,0.3m L·m in-1;inject volume,2μL.The other settingsare:scanningmode,anionmode;ion source,electrospray ionization(ESI).Theactualmass is to beadded by one proton unit (1.0078).
The1H NMR experimentswere performed with the(400-MHz) BrukerAVANCE III400NMR spectrometer.The sampleswere prepared as follows.A liquots of catalysis(100 m L in 500-m L Erlenmeyer flasks)were prepared using 50μg·mL-1catecholand 0.2mg·mL-1laccase.A fter 30m in procedure reactions in an orbital shakermaintained at200 r·m in-1and 30°C,productswere extracted 3 timeswith the same volumeof precooled ethyl acetate (4°C).Then the products were purified by silica gel column chromatography and C-18 reversed-phase chromatography.Extractswereevaporated to dryness using an Eppendorf Vacufuge (Eppendorf Scientific,Westbury,NY)at30°C.The sam plew as then subjected to1H NMR analysis.
3 Resu lts and discussion
Generally,theoxidase-catalyzed reactionsarealways involved with the short-lived free radicalsduring the cleavage and re-arrangement of the activated bonds22-24.To trap these transient radical species,the reactions are stopped either by the in situ sw iftly freezing in cryogenic liquid nitrogen(77K),orby roomtemperature spin trapping using DMPO.The latterallows structurally configuration of the radical species through the isotropic hyperfine constants(hfcs,indicated by a)ofα-nitrogen andβhydrogen(i.e.,aNand aHβ)25,26.

Scheme2 DMPO trapping reaction
Fig.2a displays the time-course changeof EPR signalstabilized by freeze trapping.A broadened and strong signalwasobserved after 5 m in of reaction,and the signal decreased dramatically when the reactionwaselongated to 15m in.This EPR signal has a symmetrical featureat g~2.004 show ing a7-G peak-to-trough width.It isassigned to the o-benzosemiquinone radical,the direct productof catecholoxidation.The continuousdecreasing in EPR signal indicates that the o-benzosem iquinone radicalwas depleted in the sequential reaction.In themeantime,Q-Tof mass spectrometry(i.e.,HPLC-MS)wasperformed to detect the products. Fig.2(b,c)shows thatsubstrate catechol(C6H6O2)was converted dom inantly to the dimer(C12H8O4and C12H10O4)and partly to the trimer(C18H12O6)(seemechanism in Scheme 3 below).Evidently, this oligomerization removes the o-benzosemiquinone radical, thereby abolishing the corresponding EPR signal(Fig.2a).Fig.2e shows a new absorption(marked by an arrow)observed at~5.8 min with identicalmoleculeweight to substrate catechol(at~2.2 min).The lag effect isascribed to itsweak polarity comparedwith thatof catechol.Under the identical condition,oligomersand the lagging signalwere notobserved in the parallelexperimentswith two isomers(m-and p-dihydroxybenzene),aswellas in the parent controlswith the absenceof either catecholor laccase(Fig.2b). Therefore this lagging signal indicates isomerization of the obenzosemiquinone radical,i.e.,itsuggests that the formed isomer isalso a radicalspecies.
W ith room-temperature spin trapping protocol,two radical specieswere trapped in situ by DMPO(Fig.3a).i)ForDMPO-·OH adduct radical,the isotropic hyperfine coupling constants(hfcs) ofα-nitrogen andβ-proton characterized by aN=aHβ=14.8G lead to the four-line(1:2:2:1)spectrum;ii)For carbon-centered DMPO adduct radical,aN=15.6G and aHβ=22.8G were obtained from the six-line hyperfine pattern.In the parallel experiments, these two transient radicalswere notobserved by replacementof catecholwith m-or p-dihydroxybenzene isomers(Fig.4).In order to clarify the origin of·OH radical,the17O isotope-labeling solvent water(H217O,~80%(atom ic fraction)17O)w as used.Experimentally,the four-line EPR signal of the DMPO-·16OH radical(16O,99.757%natureabundance)was replaced by aweaker signal,due to the further hyperfine splitting of17O(I=5/2)of DMPO-·17OH radical(aN=aHβ=14.8 G,and a17O=4.6 G). Moreover,removal of the DMPO-·16OH radical signal by17O isotope unambiguously demonstrated that the other oxygencentered radicals(e.g.,peroxide,acyl,aroylor alkoxyl radicals) were not formed.Notably,the o-benzosem iquinone radical could not be trapped by DMPO.Obviously,solventwaterwas a substrateupon theoxidative reaction,which hasnotbeen reported sofar.Simultaneously,the six-line carbon-centered signal w as prom inently protruded because of the elim ination of the DMPO-·16OH radical after adding17O-labeled water.The remaining question liesw ith w hether the carbon-centered radical is aromatic or non-aromatic speciesoriginating from catecholoxidation?We ruleout the formerwhich gives aNand aHβvalues larger than thoseof the latter25,26.Since theotheroxygen-centered radicalswere notobserved experimentally,these two radicalswere probably not released from the intra-(atC1-C2site)or extra-diol (atC1-C6site)cleavageswhich was sim ilar to those in catalysisby catecholdioxygenaseor in chemical oxidation20,21,27-32.Therefore, the trapped non-aromatic carbon-centered radicalwas assigned to the isomer of the o-benzosem iquinone radical,which was also detected in HPLC-MS(Fig.2e).To collect the intermediates,the catalyzed reaction was stopped after 30m in by adding ethyl acetate forextraction.The crude extraction by chromatographywas subjected to1H-NMRmeasurement in d6-DMSO.Fig.3b shows thata new group of1H signal in the extraction wasappeared atδ~6.0 at theexpensed of those(δ~6.7)of substrate catechol,and this novel1H signalwasassigned to the oxole derivant(i.e.,the furan derivative)33-36.
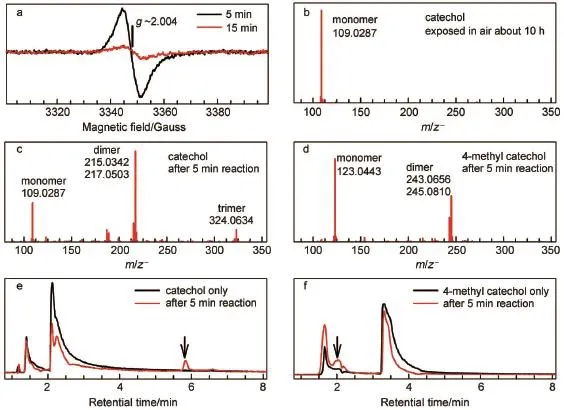
Fig.2(a)X-band(9.39GHz)EPR spectra recorded tomonitor themetastable radicalsupon the laccase-catalyzed oxidation of catechol trapped by freezing trapping;(b-d)an ion-m ode HPLC-M S from con trolsubstrate self-oxidized atam bient condition; (e,f)corresponding HPLC-MSspectra from catechol/4-methylcatecholand after 5min of reaction w ith laccase

Scheme 3 Proposedmechanism of the laccase-catalyzed extra-and intra-molecular adductsof catechol

Fig.3(a)Room tem perature X-band(9.86GHz)EPR spectra and simu lation of DMPO adduct radicalsafter 5m in of laccase-catalyzed oxidation in the normal(H216O)or17O-labeled(H217O,~80%(atom ic fraction)17O)aqueoussolution; (b)compared1H-NMR spectra of substrate catechol(black)and product(red)
For the uncoupled o-benzosem iquinone radical,the isotropic hfcs of aH4,5=3.56G and aH3,6=0.9G are known for two sets of two equivalentprotons18,24.The larger hfc of aH4,5indicates thata large positive spin density is localized atC4,5and it isnegativeat H4,518,24.Under the self-oxidized conditions,the oligomeric or adductive productswerenotdetected.Obviously,the formed obenzosem iquinone radical alone does not yield any adduct product.When adding laccase,a chelate complex was formed between the o-benzosemiquinone radicaland thecounterion Cu1+/ Cu2+(T1)19-21,27-32.The inductive effect caused by Cu ion activates the cation radical tendency at the C4-C5site.This isproved by the dimer and trimmer formed efficiently from the inter-molecular adducton the C4-C5site37,38.In particular,the trapped non-aromatic carbon-centered radical is probably an isomer derived from the intramolecular adduct of the o-benzosem iquinone radical.To test the hypothesis,analogues of catecholwere surveyed comparatively.In Fig.4,when isomers(m-and p-dihydroxybenzene)were oxidized by laccase,neither the radicals trapped by DMPO nor the oligomerswere observed,aswellas3-or4-nitroso catechol,and 2,3-dihydroxynaphthalene.However,formations of DMPO-trapped radicalsand dimerizationwerenotaffected in the presence of 3-or4-methyl catechol(Fig.1(d,f)and Fig.4).Again,noneof theotheroxygen-centered radicalswas observed in the parallel experiments.
To describe the laccase-catalyzed inter-and intra-molecular adductsof catechol,amechanism isproposed in Scheme 3,w ith comparison to the well-established mechanism of catechol dioxygenase20,21,27-30.Upon catalysis,Cu2+(T1)is reduced firstly to the metastable Cu1+(T1)and catechol is oxidized to the o-benzosem iquinone radical.Chem ical turnover from Cu1+(T1)to Cu2+(T1) isaccomplished after delivering an electron to the downstream.At T2/T3sites,O2is reduced to H2O after accepting four electrons from T1(Fig.1).Interaction between the incipient o-benzosem iquinone radical and the counterion Cu2+(T1)yields a chelate intermediateas b in Scheme3.Cu2+in the chelate induces further spin density transfer such that thenascentcation radical character at the C4-C5site is fulfilled actively.The intermolecularaddition reaction from c to d is themajor pathwaywhich can be regarded as themimic bio-synthesis of biologicalmacromolecules,e.g., lignin.Inevitably,the C4―C5bond is also attacked by the intramolecularhydroxylgroup during the transition from e to g via f (depicted as theminor pathway in Scheme3).The intermediates f and g w ere characterized by EPR and1H-NMR respectively (Fig.3).The intramolecularadductgives to the sidechain carboncentered furan-derivative radicalw hen the cleavage of C4―C5bond occurs.This ring-opening is accomplishedw ith synergy of substrate water that is ascertained when the origin of hydroxyl radical is identified by17O-labeling water(H217O).Simultaneously, Cu2+(T1)is reduced to Cu1+(T1)onceagain.Tofurther determine the inductive effect of spin density transfer,some derivatives of catechol are applied as substrates.Formations of the DMPO-trapped radicalsand dimer are notaffected in the presenceof 3-or4-methyl catecholsubstrates(methylgroup causing the electron-donating inductive effects).Contrarily,these reactions are prohibited in the presence of 3-or 4-nitro catechol(―NO2group causing theelectron-withdrawing inductiveeffects),aswell as in the presenceof 2,3-dihydroxynaphthalene.When formation of the chelate com plex is prevented in the presence of m-and p-dihydroxybenzene,all these resultsarenotobserved anymore.

Fig.4 Room tem perature EPR spectra ofDMPO spin trapping in the p resenceofa variety of substrate catecholanalogs:m-and p-dihydroxybenzene,3-or 4-methyl catecholsubstrate,3-or 4-nitroso catechol,and 2,3-dihydroxynaphthalene
Thismechanism rem indsus of another oxidationmechanism of catechol catalyzed by dioxygenase.Although the protein ligation to Cu2+(T1)ismore or lesssimilar to thataround Fe2+in catechol dioxygenase19-21,the inductive effectsof spin density transfer on the substrate oxidation are distinguished from each other.For dioxygenase,catecholand O2substratesbinding and ring-opening arecarried outsimultaneously in adjacentposition19-21.The distinct catalyticmechanismsof catechol dioxygenaseand laccase provide the first-hand evidence for further understanding and utilization of Fe/Cu co-catalysis and synergy39.These comparative results show that the radical transformation can be m im icked if the electronic structurebetween substrate intermediate(s)and catalytic ion center can be adjusted biologically or chem ically.In addition to the aromatic building block for oligomerization,the transient furan-derivative radicalmighthave potentialapplication in flavor chemistry,or phenols′antioxidation and interaction ofmulticopper oxidase in vivo with degenerative diseasessuch as cancer,heart disease,inflammation and even ageing18.This also sheds lighton the further comprehension of laccase-catalyzed transformation in PAHsbiodegradation,green chemistry,and flavor industry.
4 Conc lusions
Phenoloxidation and transformation catalyzed by laccasewas studiedmechanistically withmass spectrometry and EPR.The data shows that the chelate interacting between Cu2+(T1)ion and o-benzosem iquinone radical plays a pivotal role to direct the subsequent inter-or intra-molecularadducts,i.e.,oligomerization or isomerization correspondingly,when the electronic structureof the chelate was altered purposely by the differentmodified catecholanalogs.The proposedmechanism ishelpful to understand the distinctcatalysisof Cu and Fe that induce differentelectron spin transfer in the short-lived intermediates.Thisalso shows that the radical transformation can be m imicked if the electronic structure between substrate intermediate(s)and catalytic ion center can be adjusted biologically or chem ically tofurtherexploit the utilization of laccase.
(1)Solomon,E.I.;Sundaram,U.M.;Machonkin,T.E.Chem.Rev. 1996,96,2563.doi:10.1021/cr950046o
(2)Solomon,E.I.;Heppner,D.E.;Johnston,E.M.;Ginsbach,J. W.;Cirera,J.;Qayyum,M.;Kieber-Emmons,M.T.;K jaergaard, C.H.;Hadt,R.G.;Tian,L.Chem.Rev.2014,114,3659. doi:10.1021/cr400327t
(3)ten Have,R.;Teunissen,P.J.M.Chem.Rev.2001,101,3397. doi:10.1021/cr000115l
(4)Jiang,B.;Xu,X.Y.;Li,Z.Y.Chin.J.Org.Chem.2008,28, 1715.[姜标,徐向亚,李祖义.有机化学,2008,28,1715.]
(5)Lee,S.K.;George,S.D.;Antholine,W.E.;Hedman,B.; Hodgson,K.O.;Solomon,E.I.J.Am.Chem.Soc.2002,124, 6180.doi:10.1021/Ja0114052.
(6)Hua,J.L.;Song,C.S.;Wang,G.H.;Cai,M.Z.Acta Phys.-Chim.Sin.1999,15,173.[花建丽,宋才生,王光辉,蔡明中.物理化学学报,1999,15,173.]doi:10.3866/PKU. WHXB19990215
(7)Ma,G.X.;Zhong,Q.D.;Lu,T.H.Sci.China Chem.2010,53, 1332.doi:10.1007/s11426-010-3186-x
(8)Zeng,H.;Liao,L.W.;Li,M.F.;Tao,Q.;Kang,J.;Chen,Y.X. Acta Phys.-Chim.Sin.2010,26,3217.[曾涵,廖铃文,李明芳,陶骞,康婧,陈艳霞.物理化学学报,2010,26, 3217.]doi:10.3866/Pku.Whxb20101208
(9)Zhang,Q.;Zhang,Y.Q.;Liu,X.Z.;Liang,Y.X.;Zhang,R.F.; Long,N.B.Chin.J.Inorg.Chem.2013,29,2065.[张群,张育淇,刘晓贞,梁云霄,张瑞丰,龙能兵.无机化学学报, 2013,29,2065.]doi:10.3969/j.issn.1001-4861.2013.00.324
(10)Li,Y.;Jiang,G.X.;Niu,J.F.;Wang,Y.;Hu,L.J.Prog.Chem. 2009,21,2028.[李阳,蒋国翔,牛军峰,王颖,呼丽娟.化学进展,2009,21,2028.]
(11)Gupta,A.;Aartsma,T.J.;Canters,G.W.J.Am.Chem.Soc. 2014,136,2707.doi:10.1021/ja411078b
(12)Drauz,K.;Gröger,H.;May,O.Enzyme Catalysis in Organic Synthesis,3rd ed.;Drauz,K.,Gröger,H.,M ay,O.Eds.;W iley-VCH:Weinheim,2012.
(13)Polaina,J.;M acCabe,A.P.IndustrialEnzymes:Structure, Function,and Applications;Springer:Dordrecht,2007.
(14)Xu,F.;Damhus,T.;Danielsen,S.;Østergaard,L.H.Catalytic Applicationsof Laccase.In Modern Biooxidation,Wiley-VCH Verlag GmbH&Co.KGaA,2007;pp 43-75.
(15)Heppner,D.E.;K jaergaard,C.H.;Solomon,E.I.J.Am.Chem. Soc.2013,135,12212.doi:10.1021/ja4064525
(16)Heppner,D.E.;Kjaergaard,C.H.;Solomon,E.I.J.Am.Chem. Soc.2014,136,17788.doi:10.1021/ja509150j.
(17)Wan,Y.Y.;Du,Y.M.;M iyakoshi,T.S.Sci.China Ser.B 2008, 51,669.doi:10.1007/s11426-008-0071-y
(18)Rappoport,Z.The Chemistry ofPheno ls;W iley:Hoboken,NJ, 2003.
(19)Weinshilboum,R.M.;Otterness,D.M.;Szum lanski,C.L. Annu.Rev.Pharmacol.Toxicol1999,39,19.doi:10.1146/ annurev.pharm tox.39.1.19
(20)Brown,C.K.;Vetting,M.W.;Earhart,C.A.;Ohlendorf,D.H. Annu.Rev.Microbiol.2004,58,555.doi:10.1146/annurev. m icro.57.030502.090927
(21)Kovaleva,E.G.;Lipscomb,J.D.Science 2007,316,453. doi:10.1126/science.1134697
(22)Zard,S.Z.Advancesin Free RadicalChemistry,Vol.2;Jai Press:Stam ford,Conn.,1999.
(23)Chatgilialoglu,C.;Studer,A.Encyclopedia ofRadicalsin Chemistry,Biology and Materials;W iley-Blackwell:Ox ford,2012.
(24)Gerson,F.;Huber,W.Electron Spin Resonance Spectroscopy of Organic Radicals;Wiley-VCH:Weinheim,GreatBritain,2003.
(25)Buettner,G.R.Free Radic Biol.Med.1987,3,259. doi:10.1016/S0891-5849(87)80033-3
(26)Alberti,A.;Macciantelli,D.Spin Trapping.In Electron Paramagnetic Resonance,John W iley&Sons,Inc.:Hoboken, New Jersey,2008;pp 285-324.
(27)Sanvoisin,J.;Langley,G.J.;Bugg,T.D.H.J.Am.Chem.Soc. 1995,117,7836.doi:10.1021/Ja00134a041
(28)Spence,E.L.;Langley,G.J.;Bugg,T.D.H.J.Am.Chem.Soc. 1996,118,8336.doi:10.1021/ja9607704
(29)Lin,G.;Reid,G.;Bugg,T.D.H.J.Am.Chem.Soc.2001,123, 5030.doi:10.1021/ja004280u
(30)Xin,M.;Bugg,T.D.H.J.Am.Chem.Soc.2008,130,10422. doi:10.1021/ja8029569
(31)Brivio,M.;Schlosrich,J.;Ahmad,M.;Tolond,C.;Bugg,T.D. H.Org.Biomol.Chem.2009,7,1368.doi:10.1039/b821559f
(32)M ander,L.N.;W illiams,C.M.Tetrahedron 2003,59,1105. doi:10.1016/S0040-4020(02)01492-8
(33)Spirkova,K.;Kada,R.Chem.Pap.1987,41,787.
(34)Babij,N.R.;M cCusker,E.O.;Whiteker,G.T.;Canturk,B.; Choy,N.;Creemer,L.C.;De Amicis,C.V.;Hew lett,N.M.; Johnson,P.L.;Knobelsdorf,J.A.;Li,F.Z.;Lorsbach,B.A.; Nugent,B.M.;Ryan,S.J.;Smith,M.R.;Yang,Q.Org. Process.Res.Dev.2016,20,661.doi:10.1021/acs. oprd.5b00417
(35)Fulmer,G.R.;Miller,A.J.M.;Sherden,N.H.;Gottlieb,H.E.; Nudelman,A.;Stoltz,B.M.;Bercaw,J.E.;Goldberg,K.I. Organometallics2010,29,2176.doi:10.1021/om100106e
(36)Gunther,H.A.Nmr Spectroscopy:Basic Principles,Concepts and Applicationsin Chemistry,3rd ed.;completely revised and updated edition.
(37)Pietruszka,J.;Wang,C.Green Chemistry 2012,14,2402. doi:10.1039/C2GC35662G
(38)Abdel-M ohsen,H.T.;Conrad,J.;Beifuss,U.J.Org.Chem. 2013,78,7986.doi:10.1021/jo401193e
(39)Su,Y.J.;Jia,W.;Jiao,N.Synthesis-Stuttgart2011,1678. doi:10.1055/s-0030-1260028
RadicalMechanism of Laccase-Catalyzed Catecho lRing-Opening
CHEN M ing1WANG Lin2TAN Tian1LUO Xue-Cai2ZHENG Zai2
YIN Ruo-Chun2SU Ji-Hu1,*DU Jiang-Feng1
(1CASKey Laboratory ofMicroscaleMagnetic Resonance,DepartmentofModern Physics, University ofScience and Technology ofChina,Hefei230026,P.R.China;
2Inspiration Centerfor Bio-manufacture,SchoolofLife Sciences,AnhuiUniversity,Hefei230601,P.R.China)
Enzyme-cata lyzed reactions are a prom inent field of research in green chem istry.Laccase is a multicopper oxidase,which we used to study the oxidation of catechol.Amechanism for this ring-opening reaction is also proposed.A o-benzosem iquinone rad icalw as the initialnascent productof catecholoxidation during the cata lytic reaction.This radicalunderwent two reaction pathways:(1)formation ofan intramolecu lar adduct,w hich gave a ca rbon-centered furan-derived rad ica l trapped by 5,5-d ime thy l-1-pyrroline-N-oxide (DMPO);(2)formation ofan intermolecu lar adduct producing dimeric and trimeric oligomers,as resolved by mass spectrometry.Products of the furan-like intermediate were also characterized by1H-NMR.Simultaneously, a hydroxyl radical(·OH)originating from the w ate r so lven twas identified by17O-isotope tracing.The kinetics of this radicalwere also evidentw ith substrates including 3-and 4-methylcatechol,butnotwith resorcino land hydroquinone isomers,3-and 4-nitro catechol,and 2,3-dihydroxynaphtha lene.Themechanism ofselectiveactivation and ring-opening at the C4-C5site is discussed.This reaction is distinct from intra-and extra-diolringcleavages catalyzed by catecho ldioxygenase.These resu lts a rem eaning ful form im icking laccase cata lysis tofurther protein design.
Laccase;Radicaloxidation;Aromatic ring c leavage;Electron paramagnetic resonance spectroscopy;Spin trapping
O643
10.3866/PKU.WHXB201612011
www.whxb.pku.edu.cn
Received:September 29,2016;Revised:December 1,2016;Published online:December 1,2016.
*Corresponding author.Email:sujihu@ustc.edu.cn;Tel:+86-551-63607672.
Theprojectwas supported by theNational Key Basic Research Program of China(973)(2013CB921802),and FundamentalResearch Funds for the Central Universities,China,State Key Laboratory for Conservation and Utilization of SubtropicalAgro-bioresources,China(KSL-CUSAb-2012-03).国家重点基础研究发展规划项目(973)(2013CB921802)和亚热带农业生物资源保护与利用国家重点实验室开放课题基金(KSL-CUSAb-2012-03)资助©Editorialoffice of Acta Physico-Chim ica Sinica
猜你喜欢
疯狂英语·初中天地(2022年2期)2022-07-07 08:50:46
疯狂英语·初中版(2022年2期)2022-05-04 13:54:49
化学分析计量(2021年6期)2021-06-22 09:12:52
闽南师范大学学报(自然科学版)(2020年1期)2020-06-05 06:19:00
生物技术通报(2019年9期)2019-09-18 10:05:32
电子测试(2018年1期)2018-04-18 11:52:24
发明与创新(2016年33期)2016-08-21 13:22:22
合成化学(2015年4期)2016-01-17 09:01:04
海军航空大学学报(2015年1期)2015-11-11 17:22:41
中共合肥市委党校学报(2014年2期)2014-08-03 05:58:26
