
微流控技术制备球形发射药及其表征
2017-05-07 01:06:39刘换敏李兆乾王彦君董朝阳裴重华
含能材料 2017年9期
刘换敏, 李兆乾, 王彦君, 董朝阳, 裴重华
(1. 西南科技大学四川省非金属复合与功能材料重点实验室——省部共建国家重点实验室培育基地, 四川 绵阳 621010; 2. 泸州北方化学工业有限公司, 四川 泸州 646003)
1 引 言
球形发射药因其装填密度高、流散性好、易于钝感、易损性低而应用于小口径轻武器。球形发射药的传统制备工艺主要有内溶法[1-2]和外溶法[3]。内溶法制球是在加热和搅拌条件下,利用界面张力直接成球形液滴,液滴固化得到微球,所得球形药的尺寸分布宽,废药率高,增加了生产成本[1-2]。为提高微球内部孔结构的均匀性,王萍等[4]对溶剂蒸馏制备硝化棉基球形药后期溶剂驱除方式进行了改进,由升温蒸馏改为水溶液常温浸析,所得产物球形度较高,表面光滑,粒径集中在20 μm。外溶法成球工艺所用溶剂少,但所得球形药的结构和成分不均匀、燃烧规律性差,影响弹道性能的稳定性,且大多需要经过挤出造粒等工序,工序复杂,生产周期长[3]。因此为了降低弹道性能的偏差,通过精密制造获得粒径均一、大小可控、组织结构均匀的球形发射药非常关键,微流控技术的问世为此提供了契机,该技术制备出的微球单分散性好、粒径均一、成分和形态可控[5]。作为一种制备微球及其他微型颗粒的新兴技术,其装置操作简单,溶剂用量少,反应时间短,易于精确控制,在液滴微反应器[6-7]、临床诊断[8]、药物运输和释放[9-10]、细胞分析[11-12]等领域应用广泛。目前,利用微流控技术已可制备包括聚合物材料在内的多种材料的微球。因此本研究拟通过微流控技术来制备球形发射药。
微流控技术的关键在于其芯片结构,常用的芯片微通道的几何结构有: T型微通道[13](T-junction),流动聚焦型[14](Flow focusing)和共轴聚焦型[15](Co-flow)和台阶式微通道[16](Terrace)。由于T型结构是最简单和最早用于研究微液滴形成条件的微通道结构,相对于其他通道结构研究较成熟,故本研究采用T型微通道装置制备球形发射药。在微流控芯片中,流体流动呈层流状态,生成液滴大小主要取决于两相流速比和分散相的溶棉比(溶剂与硝化棉的质量比,简称S/N),水相与油相流速比对微球表观形貌、粒径大小的影响。
2 实验部分
2.1 试剂与仪器
试剂: 乙酸乙酯,分析纯,成都市联合化工试剂研究所; D级硝化棉(NC),工业级,泸州北方化学工业有限公司。
主要仪器: 30 mL注射器,河南曙光健士医疗器械集团股份有限公司; 数字控制注射泵(LSP02-1B),保定兰格恒流泵有限公司; 日立公司TM-1000扫描电子显微镜; 北京中科科仪有限公司SBC-12小型离子溅射仪; 马尔文2000激光粒度分析仪。
2.2 实验过程
2.2.1 微流控芯片的制备
取1片载玻片,在载玻片上制作T型通道; 分散相通道、连续相通道、输出通道分别采用内径为300,500,800 μm的塑料管; 将聚二甲基硅氧烷(PDMS)预聚体和固化剂按一定质量比混合均匀; 把混合均匀的PDMS浇铸到组合好的载玻片上; 将浇铸好的载玻片在室温条件下静置4 h,待混合胶状物中没有明显的小气泡时,在60 ℃条件下加热固化。固化成型后,取出模塑用管路,形成简易的T型通道。
2.2.2 球形发射药的制备
实验采用的是T型微通道装置,如图1所示。装置的输入端有两相: 分散相(dispersed phase)和连续相(continuous phase),分散相为NC的乙酸乙酯溶液,即油相,连续相为水,即水相,该两相分别由两台实验室注射泵控制。首先启动控制连续相的注射泵,将注射器中的连续相溶液推入到微流控主通道内。同时启动控制分散相的注射泵,将注射器中的乙酸乙酯溶液推入到微流控芯片的油相输入通道,直至末端。两相溶液在分散相输入通道末端汇合,形成油-水界面,油相末端液体受到油-水界面张力和水相剪切力的共同作用。水相提供均匀的剪切力促使油相液体脱离输入管末端,形成独立的NC液滴,而两相的界面张力有助于NC液滴保持球形,以维持NC液滴内部的稳定性。随着注射泵不断推进,形成的NC液滴随着连续相不断前进,NC液滴在前进的过程中,在界面张力的作用下,趋于球形,同时,NC液滴也逐渐固化,待液滴固化9~15 min,沉积到收集装置底部。待液滴固化沉积完全,去除上部溶液及杂质,得到的底部固体颗粒即为目标产物。整个实验过程在常温下进行。
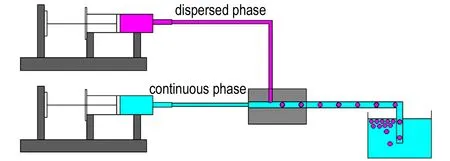
图1 微流控技术制备微球的装置示意图
Fig.1 Schematic of microfluidic device for preparing microspheres
3 结果与讨论
3.1 两相流速比对微球的影响
固定溶棉比为50∶2.5,连续相流速(Qc)为1000 μL·min-1,分散相流速(Qd)依次设定为30,50,100 μL·min-1。分别在这三种条件下利用微流控装置制备球形发射药,在实验过程中观察到,制得的液滴以滴流[17]方式生成且均匀稳定; 当分散相流速提高到100 μL·min-1以上时,由于连续相对分散相的挤压和剪切作用减小,此时液滴脱离分散相变得困难,液滴生成方式由滴流转变为射流[17],射流状态下生成的液滴不稳定。对所得样品进行SEM测试,观察不同流速比条件下的球形药形貌特征,如图2所示。从图2中观察到所得球形药的单分散性很好,且随着分散相流速增大,球形药的粒径也随之增大,其粒径从270 μm增大至306 μm。
分析图2中不同流速比制备的硝化棉球形药,结果显示溶棉比为50∶2.5,两相相流速比为1000 μL·min-1∶100 μL·min-1时,制得的硝化棉球形药的效果最好。故对该条件下制备的硝化棉球形药及其内部结构进行SEM测试,结果如图3所示。从图3中观察到硝化棉球形药的表面气孔很小、比较光滑,而且其内部结构比较密实、组织结构均匀。

a.Qc∶Qd=1000∶30 b.Qc∶Qd=1000∶50 c.Qc∶Qd=1000∶100
图2 不同流速比下制得的硝化棉球形药的SEM图
Fig.2 SEM images of nitrocellulose spheres at different flow rate ratios

a. nitrocellulose sphere b. inner structure
图3 硝化棉球形药及内部结构的SEM图
Fig.3 SEM images of nitrocellulose sphere and inner structure
采用激光粒度分析仪对溶棉比为50∶2.5,两相相流速比为1000 μL·min-1: 100 μL·min-1时所得硝化棉球形药进行粒径测试,测得的粒度分布如图4所示。激光粒度仪可测量的粒度范围为0.02~2000 μm,在测试大颗粒(大于100 μm)时会存在一定的系统误差,但测得的中位径仍具有一定的参考性,由此可以定性地分析粒径的大小及其分布状况。测试结果显示,硝化棉球形药单分散性好,球形药的粒径集中在约306 μm处。
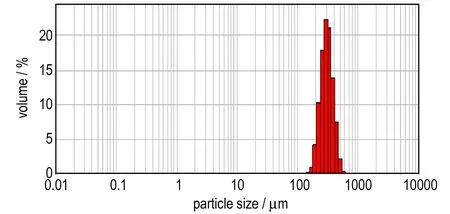
图4 硝化棉球形药粒径分布
Fig.4 Particle size distribution of nitrocellulose spheres
3.2 溶棉比(S/N)对球形药粒径的影响
固定分散相和连续相的流速比,通过调节分散相的溶棉比制备出了不同粒径的硝化棉球形药。连续相和分散相均用30 mL注射器盛装,控制水油两相流速比为1000 μL·min-1: 100 μL·min-1。连续相组分固定不变,随着溶棉比的增大,所得球形药的粒径减小。本实验制备出的球形发射药粒径集中在250~350 μm之间,并且其表观形貌规整,粒径分布窄,单分散性好(图5)。固定分散相和连续相的流速,溶棉比(S/N)为50∶3.0时,可以制备出粒径为350 μm左右的硝化棉球形药; 溶棉比为50∶2.0时,制备出的硝化棉球形药粒径约250 μm。固定两相流速比,在微通道尺寸固定不变时,溶棉比为50∶3.0~50∶2.0范围内呈滴流状生成液滴,此时液滴均匀,固化得到的球形药粒径分布窄,且溶棉比越小,所得球形药粒径越大; 当溶棉比在50∶3.0以下时,在剪切力的作用下,液滴较难脱离分散相,液滴以射流状生成,此时所得液滴均匀性差,固化得到的球形药表面略粗糙、粒径分布宽。因此,在一定范围内通过调节分散相的溶棉比可控制硝化棉球形药的粒径。
3.3 溶棉比(S/N)对球形药堆积密度(ρb)的影响
固定连续相和分散相的流速比为1000 μL·min-1∶100 μL·min-1,连续相组分固定不变,通过调节分散相的溶棉比制备出了不同粒径的硝化棉球形药。测试不同溶棉比下制得的球形硝化棉的堆积密度,溶棉比为50∶2.0,50∶2.5,50∶3.0,50∶3.5,50∶4.0时,其密度依次为0.77,0.80,0.81,0.80,0.78 g·cm-1。溶棉比为50∶2.5~50∶3.5时,硝化棉球形药的堆积密度呈上升趋势,是因为此时,球形药粒径变化不大,约200 μm,如图5a与图5b所示。此时,随分散相中硝化棉含量增多,球形药密度随之增大; 而当溶棉比为50∶4.0时,球形药粒径增大,达350 μm,如图5c所示,此时球体堆积时,相互之间的空隙增大,导致堆积密度下降。因此,分散相的溶棉比为50∶2.5~50∶3.5时可制得较高堆积密度的硝化棉球形药。

a.S/N=50∶2.0 b.S/N=50∶3.0 c.S/N=50∶4.0
图5 不同溶棉比下制得的硝化棉球形药的SEM图
Fig.5 SEM images of nitrocellulose spheres at differentS/N
4 结 论
(1)利用T型微通道装置精确控制液滴的生成,成功地制备了单分散性好的百微米级球形发射药,且所得球形药的表观形貌规整,粒径分布窄,单分散性好。
(2)固定连续相流速为1000 μL·min-1,在微通道尺寸固定不变,两相的驱动压力接近时,分散相流速和分散相的溶棉比影响球形药大小。水油两相流速比为1000 μL·min-1∶100 μL·min-1,溶棉比为50∶3.0时,液滴生成高度分散、均一稳定,且固化所得的球形发射药粒径最大且分散性好、粒径均一、球形度高。固定分散相溶棉比为50∶2.5,分散相流速由30 μL·min-1增大到100 μL·min-1过程中,所得微球的粒径从270 μm增大至306 μm,微球外观形貌由不规则逐渐变得规则均匀; 固定水油两相流速比为1000 μL·min-1∶100 μL·min-1,溶棉比从50∶3.0增大到50∶2.0,微球粒径从350μm减小到250 μm,微球表观形貌光滑。
(3)水油两相流速比为1000 μL·min-1∶100 μL·min-1,溶棉比在50∶2.5~50∶3.5时,生成的液滴高度分散、均一稳定,且所得的球形发射药的堆积密度在溶棉比为50∶3.0时最高。故在微通道尺寸固定不变时,获得高密度、粒径均一、单分散性好的球形药的最佳溶棉比为50∶3.0。
参考文献:
[1] Cox C D, Liggett T. Spherical production of small particle nitrocellulose: US, US3671515[P]. 1972.
[2] Fung K F. High productivity spheroid nitrocellulose: US, US5144020[P]. 1992.
[3] Ohtate E, Higuchi A. Nitrocellulose composition and process for producing the same: US, US4460411[P]. 1984.
[4] 王萍, 张磊, 蔺向阳, 等. 溶剂浸析法制备硝化棉基微孔球形药[J]. 含能材料, 2015, 23(11): 1107-1110.
WANG Ping, ZHANG Lei, LIN Xiang-yang, et al. Preparation of nitrocellulose-based micro-pores spherical powder by solvent leaching method[J].ChineseJournalofEnergeticMaterials(HannengCailiao), 2015, 23(11): 1107-1110.
[5] 林炳承. 纳米科学与技术:微纳流控芯片实验室[M]. 第一版. 北京:科学出版社, 2015: 153-182.
LIN Bing-cheng. Nanoscience and Nanotechnology: Lab on a Microfluidic Chip[M]. The First Edition. Beijing: Science Press, 2015: 153-182.
[6] Xu J, Xu X, Zhao H, et al. Microfluidic preparation of chitosan microspheres with enhanced adsorption performance of copper(Ⅱ)[J].Sensors&ActuatorsBChemical, 2013, 183(13): 201-210.
[7] Zamoramora V, Velasco D, Hernández R, et al. Chitosan/agarose hydrogels: cooperative properties and microfluidic preparation[J].CarbohydratePolymers, 2014, 111(1): 348-355.
[8] Wei X, Tian T, Jia S, et al. Target-responsive DNA hydrogel mediated “stop-flow” microfluidic paper-based analytic device for rapid, portable and visual detection of multiple targets[J].AnalyticalChemistry, 2015, 87(8): 4275-4282.
[9] Pessi J, Santos H A, Miroshnyk I, et al. Microfluidics-assisted engineering of polymeric microcapsules with high encapsulation efficiency for protein drug delivery[J].InternationalJournalofPharmaceutics, 2014, 472(1-2): 82-87.
[10] Kong F, Zhang X, Hai M. Microfluidics fabrication of monodisperse biocompatible phospholipid vesicles for encapsulation and delivery of hydrophilic drug or active compound[J].Langmuir, 2014, 30(13): 3905-3912.
[11] Gao D, Wei H, Guo G S, et al. Microfluidic cell culture and metabolism detection with electrospray ionization quadrupole time-of-flight mass spectrometer[J].AnalyticalChemistry, 2010, 82(13): 5679-5685.
[12] Wang B L, Ghaderi A, Zhou H, et al. Microfluidic high-throughput culturing of single cells for selection based on extracellular metabolite production or consumption[J].NatureBiotechnology, 2014, 32(5): 473-478.
[13] Janhn A, Reiner J E, Vreeland W N, et al. Preparation of nanoparticles by continuous-flow microfluidics[J].JournalofNanoparticleResearch, 2008, 10(6): 925-934.
[14] Zhang S, Yun J, Shen S, et al. Formation of solid lipid nanoparticles in a microchannel system with a cross-shaped junction[J].ChemicalEngineeringScience, 2008, 63(23): 5600-5605.
[15] Baroud C N, Gallaire F, Dangla R. Dynamics of microfluidic droplets[J].LabonaChip, 2010, 10(16): 2032-2045.
[16] Sugiura S, Nakajima M, Seki M, et al. Prediction of droplet diameter for microchannel emulsification[J].Langmuir, 2002, 18(10): 3854-3859.
[17] 刘志鹏, 徐进良, 张伟. T型微通道内液滴流型分布及不稳定性分析[J]. 微纳电子技术, 2008, 45(5): 275-281.
LIU Zhi-peng, XU Jin-liang, ZHANG Wei. Flow patterns and instabilities of monodisperse droplet formation in a microfluidic T-junction device[J].MicronanoelectronicTechnology, 2008, 45(5): 275-281.
猜你喜欢
建材发展导向(2021年24期)2021-02-12 02:00:02
中国粉体技术(2021年1期)2021-01-04 02:19:28
东华大学学报(自然科学版)(2018年1期)2018-06-29 03:35:10
中国塑料(2016年6期)2016-06-27 06:34:18
中国塑料(2016年4期)2016-06-27 06:33:40
分析测试学报(2015年4期)2016-01-13 06:18:34
分析测试学报(2015年3期)2016-01-13 06:18:11
分析测试学报(2015年3期)2016-01-13 06:18:10
合成纤维工业(2015年4期)2015-03-25 12:52:38
橡胶工业(2015年8期)2015-02-23 23:41:15
