
敲除pknG提高谷氨酸棒杆菌L-谷氨酸和GABA产量
2017-04-30 03:26:59王楠楠倪亚兰
食品与生物技术学报 2017年2期
王楠楠, 倪亚兰, 史 锋*
(1.食品科学与技术国家重点实验室,江南大学,江苏 无锡 214122;2.江南大学 工业生物技术教育部重点实验室,江苏 无锡214122;3.江南大学 食品安全与营养协同创新中心,江苏 无锡214122)
敲除pknG提高谷氨酸棒杆菌L-谷氨酸和GABA产量
王楠楠1,2,3, 倪亚兰1,2,3, 史 锋*1,2,3
(1.食品科学与技术国家重点实验室,江南大学,江苏 无锡 214122;2.江南大学 工业生物技术教育部重点实验室,江苏 无锡214122;3.江南大学 食品安全与营养协同创新中心,江苏 无锡214122)
γ-氨基丁酸(GABA)具有多种生理功能,被广泛应用于食品、医药等行业。在谷氨酸棒杆菌ATCC13032中表达来自短乳杆菌的两个谷氨酸脱羧酶(GAD)编码基因gadB1、gadB2,可将其自身积累的L-谷氨酸有效转变成GABA。为了进一步提高GABA产量,首先在ATCC13032中敲除了丝氨酸/苏氨酸蛋白激酶PknG的编码基因pknG,以提高GABA的前体物质L-谷氨酸的供应,然后将GAD表达质粒pJYW-4-gadB1-gadB2转入pknG敲除菌株和ATCC13032中,构建出重组菌SNW203和SNW200,最后对两个重组菌进行上罐发酵。结果表明:相对于SNW200,菌株SNW203的L-谷氨酸和GABA产量都有所提高。发酵72 h后,GABA产量为(30.2±0.3)g/L,相对于SNW200提高了55.4%,L-谷氨酸和GABA的总摩尔浓度为0.3 mol/L,提高了36.4%。这说明敲除pknG能够促进重组谷氨酸棒杆菌的L-谷氨酸和GABA生物合成。
γ-氨基丁酸;谷氨酸棒杆菌;pknG基因
γ-氨基丁酸(γ-aminobutyric acid,GABA)是一种非蛋白质氨基酸,是哺乳动物中枢神经系统一种重要的抑制性神经递质,具有降血压、镇静安神[1]、健肝利肾、调节激素分泌[2]、抑制癌症等功能。因此,GABA作为一种功能性因子被广泛应用于食品、医药等行业。GABA是由谷氨酸脱羧酶(Glutamate decarboxylase,GAD)催化L-谷氨酸发生α-脱羧反应生成的。几种细菌GAD基因已被确定,比如大肠杆菌 (Escherichia coli)和乳酸菌 (Lactic acid bacteria,LAB)。LAB是对人类和动物有益的益生菌群,因此是最适合生产GABA的微生物[3]。然而,通过LAB生产GABA需要添加L-谷氨酸作为前体物质,并且LAB的培养需要昂贵的氮源。
谷氨酸棒杆菌 (Corynebacterium glutamicum),是一种好氧型的、非致病革兰氏阳性菌,它是一种重要的工业微生物,用来发酵生产L-谷氨酸和其它氨基酸[4],及其它各种有机酸[5]。C.glutamicum中不存在GAD基因,在简单的培养基中就能生长,相对于LAB的培养,成本很低。本课题组前期在C.glutamicum ATCC13032共表达了两个GAD基因(gadB1和gadB2),摇瓶发酵84 h时,GABA产量达到了(18.7±2.1)g/L[6]。L-谷氨酸转化为GABA的转化率达到了0.60 mol/mol。为了进一步提高GABA的产量,作为GABA的唯一的前体物质L-谷氨酸的合成必须加强。
C.glutamicum中,L-谷氨酸是由谷氨酸脱氢酶(Glutamate dehydrogenase,GDH)催化α-酮戊二酸(α-Ketoglutarate,α-KG)直接合成的,但是α-酮戊二酸脱氢酶复合体 (α-Ketoglutarate dehydrogenase complex,ODHC)也会催化α-KG生成琥珀酰辅酶A,因此减少了L-谷氨酸合成的代谢流。而pknG会影响ODHC的活性,具体作用机制如图1。

图1 ODHC的亚基组成和OdhI、PknG对ODHC活性的调控Fig.1 ODHC subunits and the regulation of ODHC activity by OdhI and PknG
ODHC由三个亚基(图1)组成:α-酮戊二酸脱氢酶 (E1亚基,OdhA)、二氢硫辛酰琥珀酰转移酶(E2亚基,AceF)、二氢硫辛酸脱氢酶(E3亚基,Lpd)[7]。OdhI(α-酮戊二酸脱氢酶抑制剂)是E1亚基OdhA的一个抑制蛋白质,非磷酸化的OdhI可以与OdhA相互作用,从而抑制ODHC的活性。而丝氨酸/苏氨酸蛋白激酶PknG催化OdhI磷酸化而使OdhI失活[8],所以敲除PknG的编码基因pknG会降低ODHC的活性,从而有利于L-谷氨酸的合成。为此对pknG基因进行敲除,研究它对重组菌GABA及其前体物质L-谷氨酸合成的影响。
1 材料与方法
1.1 材料
1.1.1 菌株、质粒和引物 本研究中用到的菌株和质粒见表1,引物见表2。
1.1.2 培养基和培养条件
1)培养E.coli时均用LB培养基(g/L):蛋白胨10,酵母膏5,氯化钠10;筛选转化子需加入30 mg/L卡那霉素。
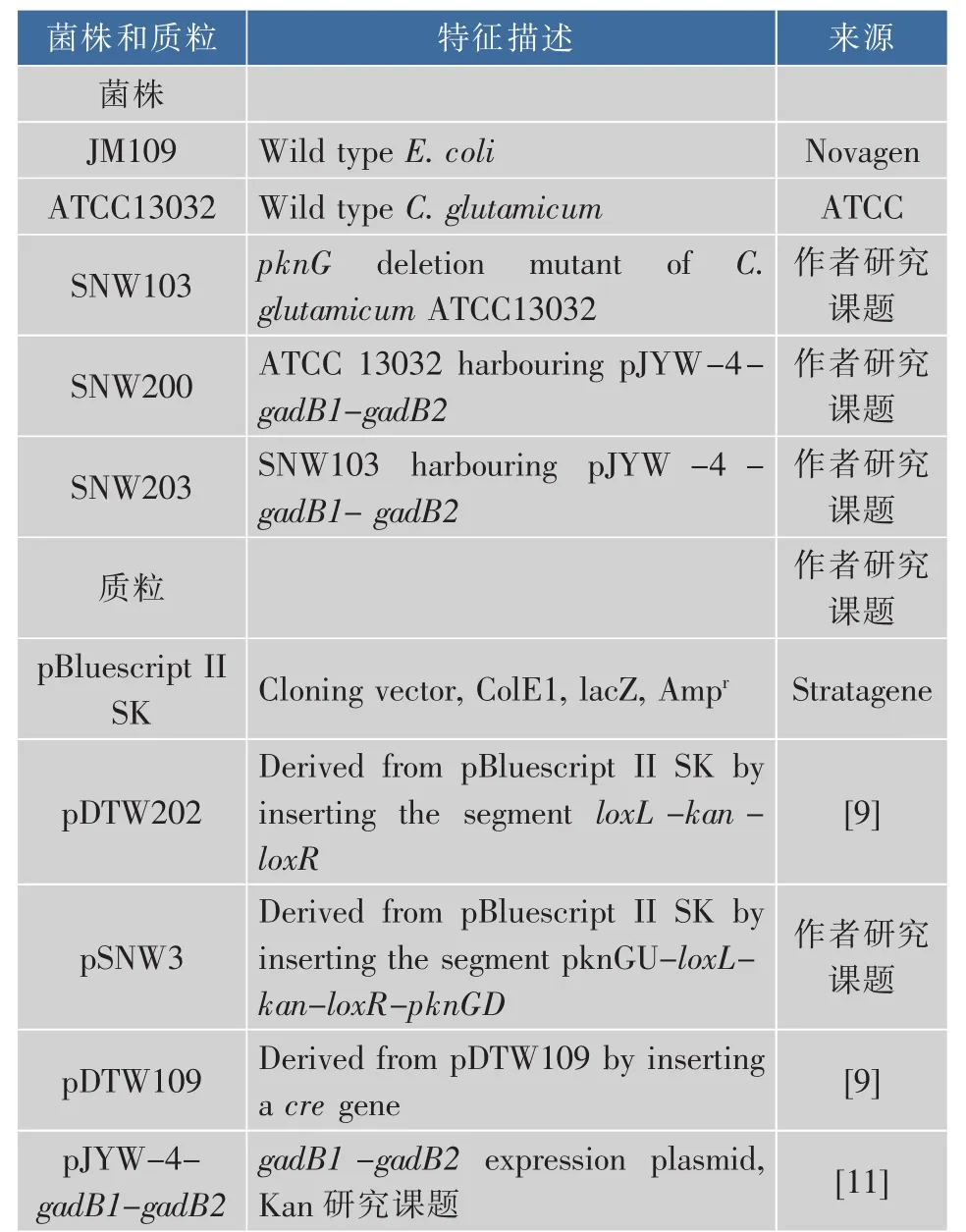
表1 本研究中所使用的菌株和质粒Table 1 Strains and plasmids in this research

表2 本研究中用到的引物Table 2 Primers in this research
2)C.glutamicum用LBG培养基(g/L):蛋白胨10,酵母粉5,氯化钠10,葡萄糖5。
3)C.glutamicum感受态培养基 (g/L):酵母粉5,蛋白胨10,NaCl 10,吐温80 1,甘氨酸30。
4)C.glutamicum电转化恢复培养基LBHIS(g/ L):酵母膏2.5,蛋白胨5,氯化钠5,脑心浸液18.5,山梨醇91;筛选转化子需加入30 mg/L卡那霉素或者15 mg/L氯霉素。
5)C.glutamicum工程菌发酵种子培养基(g/L):葡萄糖 25,玉米浆 30,尿素 8,K2HPO4·3H2O 1,MgSO40.2;卡那霉素30 mg/L;pH 7.2。
6)C.glutamicum工程菌发酵培养基(g/L):葡萄糖100,玉米浆2,尿素4,K2HPO4·3H2O 2,MgSO40.4,MnSO4·7H2O 0.2,FeSO4·7H2O 0.29;卡那霉素30 mg/L,pH 7.2。
1.1.3 工具酶和主要试剂 各种限制性内切酶,rTaq DNA聚合酶,T4 DNA连接酶,DNA Ladder Marker,购自Fermentas公司公司;核酸染料GoldView,购于上海赛百盛基因技术有限公司;OPA,GABA标样,购于Sigma公司;蛋白胨,酵母膏,脑心浸液,购于英国OXOID公司;色谱纯乙腈,甲醇,0.22 μm有机相和水相滤膜,购于上海安普科学仪器有限公司;其它试剂均购于国药集团上海化学试剂有限公司。
1.2 方法
1.2.1 pknG敲除菌株的构建 基因的敲除采用谭延振[12]和Hu等[9]报道的方法,原理是基于同源重组和位点特异性重组。首先,在E.coli JM109中构建pknG基因的敲除质粒,再转入 C.glutamicum ATCC13032中,使其发生同源重组从而敲除目的基因,然后转入携带cre基因的质粒使位于kan片段两端的lox位点发生位点特异性重组,从而去除kan片段,最后去除携带cre的质粒,进而得到pknG敲除菌株。E.coli JM109和C.glutamicum感受态细胞制备方法和转化方法参照Xu等[13]报道的方法。
1)pknG敲除质粒的构建过程:pknG敲除质粒的构建过程如图2所示:首先,以C.glutamicum ATCC13032的基因组为模板,pknGU-F/pknGU-R(5’-3’端引入的酶切位点是:Hind III和Xho I)和pknGD-F/pknGD-R(酶切位点是:Xba I和Kpn I)这两对引物,分别PCR扩增出pknG基因的上游同源臂片段pknGU和下游同源臂片段pknGD。其次,以质粒pDTW-202为模板,kanL-F/kanL-R(酶切位点是:Xho I和Xba I)为引物,扩增出kan基因片段,片段两端含有被Cre酶识别的lox位点。最后将pknG基因的上下游片段、kan片段和相应酶切过的pBluescript II SK的质粒连接,得到的新质粒即是pknG基因敲除质粒,命名为pSNW3。
2)pknG敲除菌株的构建过程:
①将敲除质粒pSNW3电转入C.glutamicumATCC13032,涂布于含有卡那霉素的恢复培养基 LBHIS平板上,30℃培养3 d左右。

图2 pknG基因敲除质粒pSNW3的构建过程Fig.2 Construction of pknG gene deletion plasmid pSNW3
②挑取平板上的转化子培养并进行PCR验证,选取验证正确的转化子,即同源重组后基因组带有kan基因片段的菌株,kan两端带有可以被Cre酶识别的lox位点。对此菌株进行感受态细胞的制备。
③为了去除基因组上的kan基因,把质粒pDTW109电转入上述感受态细胞中,此质粒是带有Cre酶的温敏型质粒,在37℃会丢失。然后涂布于含有氯霉素的恢复培养基LBHIS平板上,25℃培养一段时间至转化子长出,使基因组上的kan两端发生位点特异性重组。
④选取平板上的转化子进行PCR验证,挑取转化子验证kan基因片段是否去除,如果kan已经去除,将其接种于液体LBG培养基,37℃过夜培养,去除质粒pDTW109。
⑤挑取过夜培养的菌液于LBG固体划线,30℃培养24 h后挑取单菌落进行PCR验证和抗性验证。PCR验证正确并且在含有卡那霉素或者氯霉素的平板上均不生长的即为pknG敲除菌株,命名为SNW103。
1.2.2 GAD表达菌株的构建 gadB1和gadB2是编码GAD的两个基因,研究表明[10],基因gadB1-gadB2的共表达比两个基因的单独表达更有利于GABA的积累。
对pknG敲除菌SNW103和野生型ATCC13032进行感受态细胞制备,将携带卡那霉素抗性基因的GAD表达质粒pJYW-4-gadB1-gadB2[11]分别转入两个菌株。构建GAD表达菌SNW103/pJYW-4-gadB1-gadB2,命名为SNW203;ATCC13032/pJYW-4-gadB1-gadB2,命名为SNW200。
1.2.3 C.glutamicum工程菌株的上罐发酵C.glutamicum工程菌株的上罐发酵方法参照Wang等[11]报道的方法。发酵周期为72 h,每隔12 h取样一次,待发酵结束后,用高压液相色谱(HPLC)测定各个时间点的L-谷氨酸和GABA的产量。
1.2.4 发酵过程中残糖和氨基酸含量测定 残糖:发酵过程中取出的发酵液离心取上清液,用ddH2O稀释100倍后直接用生物传感分析仪测定葡萄糖的质量浓度。
氨基酸含量测定:氨基酸含量测定方法是邻苯二甲醛(OPA)自动柱前衍生化法[10]。
2 结果与讨论
2.1 敲除pknG的GAD菌的构建
首先,构建pknG敲除质粒pSNW3,构建过程如1.2.1所述。随后将pknG敲除质粒pSNW3转入C.glutamicum ATCC13032进行同源重组,使抗性标记基因kan与pknG基因进行替换。然后通过Cre酶识别的lox位点进行位点特异性重组,去除抗性标记基因kan,得到pknG敲除菌株SNW103。构建过程如1.2.1所述。对构建得到的敲除菌株基因组进行PCR验证,以GU/GD(上游同源臂和下游同源臂中间位置设计的引物)为引物,产物大小为1 000 bp左右,如图3的1泳道。图3的2泳道是对野生型ATCC13032基因组PCR得到的产物,大小为2 581 bp,是pknG基因未敲除的大小。对比可以得出,pknG基因敲除成功。

图3 敲除菌株SNW103和野生型ATCC13032基因组PCR验证电泳图Fig.3 Verification of the deletion strain SNW103 and wild type ATCC13032 by PCR
最后将GAD共表达质粒转入pknG敲除菌株SNW103和ATCC13032中,得到pknG敲除GAD菌SNW203和野生型GAD菌SNW200。
2.2 pknG敲除GAD菌SNW203和野生型GAD菌SNW200的上罐发酵
分别对菌株SNW203和SNW200进行上罐发酵。
2.2.1 发酵过程的控制 在L-谷氨酸和GABA合成过程中,GDH(最适pH为7.5)和GAD(最适pH为4.5)是非常重要的两个酶。为了使这两个酶发挥活性,必须将发酵过程控制为两个不同pH阶段。0~36 h,L-谷氨酸发酵阶段;36~72 h,GABA合成阶段。
在发酵前期0~36 h,发挥作用的主要是GDH,L-谷氨酸大量合成。在发酵起始时,添加4 g/L的尿素作为氮源来维持菌体的生长,这时,pH会上升,所以流加2 mol/L HCl来控制pH值在7.0~7.5之间;当pH开始下降时,流加质量浓度10 g/dL尿素控制pH值在7.0~7.5之间,同时提供了L-谷氨酸合成时所需要的氮源。
在发酵后期36~72 h,发挥作用的主要是GAD,GABA大量合成。36 h之后,停止添加尿素,L-谷氨酸的积累量达到了很高,这时,pH会下降。当pH降到适合GAD发挥活性的值时,GAD开始发挥作用,L-谷氨酸开始转化成GABA,这时pH又会升高。要使GAD充分发挥活性,pH要维持在5.0~5.5之间,所以流加2 mol/L HCl来控制pH值。
发酵周期72 h,种子转接体积分数是8%,发酵在装有1.2 L发酵培养基的3 L NBS罐中进行,温度30℃,溶氧控在体积分数30%,转速和溶氧偶联。每隔12 h取样,分别测定OD562、残糖、L-谷氨酸和GABA产量。
2.2.2 发酵过程中OD562、残糖以及耗糖的变化 发酵过程中,菌株SNW203和SNW200的OD562变化情况如图4所示。0~12 h是菌体快速生长的时期,24 h之后缓慢增长,36~72 h基本平稳,但稍有下降。发酵72 h结束时,SNW203的OD562稍高于菌株SNW200,说明pknG基因敲除后没有影响菌体的生长。

图4 SNW203和SNW200的生长Fig.4 Growth of SNW203 and SNW200 cells
菌株SNW203和SNW200发酵过程中的残糖和耗糖情况见图5。

图5 菌株SNW203和SNW200的残糖和耗糖变化Fig.5 Residual glucose and glucose consumption of SNW203 and SNW200
从残糖变化来看,0~24 h,葡萄糖被快速消耗,此过程是菌体快速生长和L-谷氨酸快速合成时期,所以葡萄糖消耗很快。24 h之后,如果残糖小于20 g/L,则需要补加葡萄糖使其维持在20 g/L以上。24~36 h,葡萄糖消耗比较缓慢,主要用于L-谷氨酸的合成。36~72 h之间,葡萄糖消耗得很少,这个过程主要是L-谷氨酸转化成GABA的过程,所以耗糖比较少。发酵72 h结束时,菌株SNW200的总耗糖为 (112.7±7.5)g/L,菌株SNW203的总耗糖为(135.1±10.2)g/L,比菌株SNW200高出19.9%。
2.2.3 发酵过程L-谷氨酸和GABA的产量以及总浓度的变化 发酵过程中,菌株SNW203和SNW200的L-谷氨酸和GABA的产量变化如图6所示。两个菌株L-谷氨酸和GABA的积累趋势是相似的,但是产量相差很大。在对照菌株SNW200中,0~36 h,L-谷氨酸大量积累,36 h到达最高值(34.7±5.8)g/L,随后缓慢下降。36 h之后,L-谷氨酸开始转化成GABA,GABA开始积累,到72 h发酵结束时,GABA的最终产量为(19.4±2.6)g/L。最终L-谷氨酸转化成GABA的摩尔转化率达到了88%。在pknG敲除GAD菌SNW203中,0~36 h,L-谷氨酸大量积累,36 h到达最高值(44.8±5.3)g/L,随后迅速下降,L-谷氨酸的最高产量比菌株SNW200的最高产量高了29.2%。36 h之后,L-谷氨酸开始转化成GABA,GABA开始积累,72 h发酵结束后,GABA的最终产量为(30.2±0.3)g/L,比SNW200高出55.4%。最终L-谷氨酸的摩尔转化率为96%。
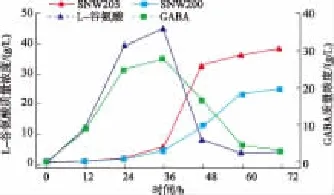
图6 菌株SNW203和SNW200的L-谷氨酸和GABA浓度变化Fig.6 L-glutamateand GABA concentrationsof SNW203 and SNW200
菌株SNW203和SNW200的L-谷氨酸和GABA的总摩尔浓度的变化如图7所示。

图7 SNW203和SNW200的L-谷氨酸和GABA总浓度变化Fig.7 Total concentration of L-glutamate and GABA of SNW203 and SNW200
在对照菌株SNW200中,0~36 h,L-谷氨酸和GABA的总摩尔浓度在不断升高,36 h达到最高值0.26 mol/L,随后有所降低,72 h发酵结束后,L-谷氨酸和GABA的总摩尔浓度是0.22 mol/L,GABA的糖酸转化率达到了0.33 mol/mol。
在pknG敲除GAD菌SNW203中,0~36 h,L-谷氨酸和GABA的总摩尔浓度在不断升高,48 h达到最高值0.32 mol/L,随后有所降低,发酵72 h结束时,L-谷氨酸和GABA的总摩尔浓度是0.30 mol/L,相对于SNW200提高了36.7%,发酵过程中的总摩尔浓度始终比SNW200高。GABA的糖酸转化率达到了0.39 mol/mol。
综上所述,敲除pknG基因后,有利于L-谷氨酸和GABA的积累。
另外,从图 6中可以看出,36~48 h,菌株SNW203的L-谷氨酸是迅速下降,而GABA是迅速升高,上升和下降的速度高于对照菌株SNW200。并且在48 h,绝大部分的L-谷氨酸已经转化成了GABA,转化速度非常快。因此在发酵过程中,可以进一步考虑缩短菌株SNW203的发酵周期,这样更有利于GABA的工业化生产。
3 结语
在C.glutamicum ATCC13032中共表达了两个GAD基因(gadB1和gadB2),可以有效地将其自身合成的L-谷氨酸转化为GABA。为了进一步提高GABA的产量,作为GABA的唯一的前体物质L-谷氨酸的合成必须加强。因此本研究中敲除pknG基因,来提高L-谷氨酸的供应,进而提高GABA的产量。首先,构建了pknG敲除菌株SNW103,然后将共表达GAD质粒转入SNW103和ATCC13032,得到pknG敲除GAD菌SNW203和对照GAD菌SNW200。
两个GAD菌株的上罐结果表明,pknG敲除后,GABA的产量从(19.4±2.6)g/L提高到(30.2±0.3)g/L,提高了55.4%。L-谷氨酸的最高产量从(34.7±5.8)g/L提高到(44.8±5.3)g/L,提高了29.2%。发酵结束后,L-谷氨酸和GABA的总摩尔浓度从0.22 mol/L提高到0.30 mol/L,提高了36.4%。可以看出,敲除pknG基因后,GABA和L-谷氨酸的产量都有所提高,pknG基因的敲除起到了很好的效果。
72 h发酵结束后,pknG敲除GAD菌SNW203的GABA产量有很大幅度的提高,L-谷氨酸摩尔转化率为96%,更有利于后期GABA的分离和纯化,为其工业化应用奠定了良好基础。
[1]WANGJiaojiao,BAIWeidong,LIANGBinxia.Thephysiologicalfunctionsandenrichmentresearchprogressofthe γ-aminobutyric acid[J].Academic Periodical of Farm Products Processing,2012(1):40-45.(in Chinese)
[2]LIN Qian.Outline of physiologic function of γ-aminobutyric acid[J].Journal of Yulin Normal University,2010,31(2):62-65.(in Chinese)
[3]LI H X,CAO Y S.Lactic acid bacterial cell factories for gamma-aminobutyric acid[J].Amino Acid,2010,39:1107-1116.
[4]TSUCHIDA T,YOSHINAGA F,KUBOTA K.Production of L-valine by 2-thiazolealanine resistant mutants derived from glutamic acid producing bacteria[J].Agric Biol Chem,1975,39:1319-1322.
[5]WENDISCH V F,BOTT M,EIKMANNS B J.Metabolic engineering of Escherichia coli and Corynebacterium glutamicum for biotechnological production of organic acids and aminoacids[J].Curr Opin Microbiol,2006(9):268-274.
[6]SHIF,JIANG J J,LIY X,et al.Enhancement ofgamma-aminobutyric acid production in recombinant Corynebacterium glutamicum by co-expressing two glutamate decarboxylase genes from Lactobacillus brevis[J].J Ind Microbiol Biotechnol,2013,40:1285-1296.
[7]KRAWCZYK S,RAASCH K,SCHULTZ C,et al.The FHA domain of OdhI interacts with the carboxyterminal 2-oxoglutarate dehydrogenase domain of OdhA in Corynebacterium glutamicum[J].FEBS Lett,2010,584:1463-1468.
[8]NIEBISCH A,KABUS A,SCHULTZ C,et al.Corynebacterial protein kinase G controls 2-oxoglutarate dehydrogenase activity via the phosphorylation status of the OdhI protein[J].J Biol Chem,2006,281:12300-12307.
[9]HU J Y,TAN Y Z,LI Y Y,et al.Construction and application of an efficient multiple-gene-deletion system in Corynebacterium glutamicum[J].Plasmid,2013,70:303-313.
[10]江君君.谷氨酸棒杆菌中共表达短乳杆菌来源的谷氨酸脱羧酶生产γ-氨基丁酸[D].无锡:江南大学生物工程学院,2013.
[11]WANG N N,NI Y L,SHI F.Deletion of odhA or pyc induce efficient γ-aminobutyric acid and its precursor L-glutamate overproduction in Corynebacterium glutamicum[J].Biotechnol Lett,2015.
[12]谭延振.谷氨酸棒状杆菌基因敲除系统的构建[D].无锡:江南大学生物工程学院,2012.
[13]XU D Q,TAN Y Z,HUAN X J,et al.Construction of novel shutter vector for use in Brevibecterium flavum,an industrial amino acid producer[J].J Microbiol Methods,2010,80:86-92.
Deletion of pknG Improves the Production of L-Glutamate and GABA in Recombinant Corynbacterium glutamicum
WANG Nannan1,2,3, NI Yalan1,2,3, SHI Feng*1,2,3
(1.State Key Laboratory of Food Science and Technology,Jiangnan University,Wuxi 214122,China;2.Key Laboratory of Industrial Biotechnology,Ministry of Education,Jiangnan University,Wuxi 214122,China;3.Synergetic Innovation Center of Food Safety and Nutrition,Jiangnan University,Wuxi 214122,China)
Gama(γ)-aminobutyric acid(GABA),which has a variety of physiological functions,is widely used in food,pharmaceutical and other industries.Two L-glutamate decarboxylase(GAD)genes (gadB1 and gadB2)were co-expressed previously in an L-glutamate producing strain Corynebacterium glutamicum ATCC13032,making the own accumulated L-glutamate be effectively transformed into GABA.To further enhance GABA production,the pknG gene encoding serine/threonine protein kinase PknG was deleted to improve the supply of GABA precursor L-glutamate.Then the co-expression plasmid pJYW-4-gadB1-gadB2 was transformed into pknG deletion strain and ATCC13032,generating the recombinant C.glutamicum strains SNW203 andSNW200.After the strains were fermented in fermenters,the production of both L-glutamate and GABA in SNW203 increased greatly when compared with SNW200.At 72 h of fermentation,GABA production in SNW203 increased to(30.2±0.3)g/L,55.4%higher than that in SNW200 and the total concentration of L-glutamate and GABA reached up to 0.3 mol/L,36.4%higher than that in SNW200.This result indicates that the deletion of pknG improves the biosynthesis of L-glutamate and GABA in recombinant C.glutamicum.
γ-aminobutyric acid,Corynbacterium glutamicum,pknG gene
Q 815
A
1673—1689(2017)02—0187—07
2015-04-16
食品科学与技术国家重点实验室自主科研课题项目(SKLF-ZZB-201405)。
*通信作者:史 锋(1970—),女,江苏丹阳人,工学博士,副教授,硕士研究生导师,主要从事微生物代谢工程与基因工程研究。
E-mail:shifeng@jiangnan.edu.cn
王楠楠,倪亚兰,史 锋.敲除pknG提高谷氨酸棒杆菌L-谷氨酸和GABA产量[J].食品与生物技术学报,2017,36(02):187-193.
猜你喜欢
河北画报(2020年10期)2020-11-26 07:20:56
英美文学研究论丛(2018年2期)2018-08-27 01:56:44
食品科学(2018年10期)2018-05-23 01:27:28
三门峡职业技术学院学报(2017年1期)2017-06-05 10:17:30
中国洗涤用品工业(2017年2期)2017-04-16 05:07:45
中国比较医学杂志(2017年5期)2017-01-17 06:17:05
西南医科大学学报(2015年1期)2015-08-22 13:01:46
医学研究杂志(2015年12期)2015-06-10 06:57:46
中国当代医药(2015年9期)2015-03-01 02:01:59
西南军医(2015年6期)2015-01-23 01:25:50
