
DNA条形码不同分析方法对鹿类动物识别效果的比较
2017-04-24 05:06马巍威刘依明董丙君杨宝田沈阳师范大学生命科学学院辽宁沈阳110034
长春师范大学学报 2017年4期
马巍威,刘依明,董丙君,杨宝田(沈阳师范大学生命科学学院,辽宁沈阳 110034)
DNA条形码不同分析方法对鹿类动物识别效果的比较
马巍威,刘依明,董丙君,杨宝田
(沈阳师范大学生命科学学院,辽宁沈阳 110034)
DNA条形码技术的任务是根据线粒体COI基因的小片段序列来识别样本到指定物种。本研究采用11种指派分析方法对8种鹿科动物30个样本序列进行分析,分别检测各分析方法的性能。结果表明,采用随机森林法和支持向量机法对鹿类动物DNA条形码分类最为可靠。没有可以适用任何情况的单一方法,对表现性能影响最大的因素是分子数据多态性。通过增加基因位点、增加测序长度和样本量可以改进多数方法的预测性能,另外,选择对分类样本最适用的分析方法可以提高分析质量。
COI;DNA条形码;鹿科动物;物种识别
DNA条形码技术旨在应用线粒体、核或质体DNA的一个较短片段作为标记,在物种水平上对现存生物类群和未知生物材料进行识别和鉴定[1]。DNA条形码具有可靠性强、成本低廉和简便易行等特性,可以在没有分类学专家参与的情况下,提供物种识别功能。线粒体细胞色素C氧化酶亚单元I(COI)是公认的动物识别DNA条形码候选基因[2]。生命条形码数据系统(Barcode of life data system,BOLD)是挖掘、储存和应用DNA条形码的全球数据库[3],截至目前,该数据库共记录偶蹄目动物210种,条形码序列3254条,其中鹿科动物40种,条形码序列804条。
DNA条形码技术能够有效鉴别和分类许多动物物种和类群[4-10],但这个目标的实现需要可靠的数学运算和分析方法。DNA条形码分析方法概括起来有基于系统树的方法(Tree-based methods)[11-13]、基于相似性的方法(Similarity-based methods)[5, 14]、基于特征(字符或碱基)的方法(Character-based methods)[15-18]、统计方法(Statistical methods)[19-21]、人工智能方法(Artificial intelligence-based methods)[22-23]和模糊集合理论方法(Fuzzy set theory-based methods)[24]。Weitschek等(2014)[25]提出监督分类法(Supervised classification methods)概念,也就是通过对已知物种的DNA条形码参考序列(reference library)分析将需要识别的未知样本序列(query set)指定到现存物种库。监督分类法包括最近邻法(1-Nearest neighbour,1-NN)、随机森林法(Random forest,RF)、基于函数的支持向量机法(Support vector machines,SVM)、内核函数法(Kernel functions)、基于规则的RIPPER法(Jrip)、决策树(Decision tree,J48)法等。
中国有18种鹿科(Cervidae)动物分布[26],它们在生物多样性和生态系统中占有非常重要的地位。受生态环境的破坏、栖息地丧失和非法捕猎等因素的影响,其野生种群数量在急剧减少,有的甚至已经灭绝,许多鹿类动物已被列为濒危物种[27]。为有效保护鹿类野生动物,促进资源的可持续发展,需要对这些动物种类及其产品进行有效的分子鉴定。由于需要分类的生物类群不同或采用的DNA条形码基因位点及序列长度不同,在物种识别能力和效果上存在一定的差异。本文选取DNA条形码11种分析方法对8种鹿科动物进行鉴别,以检测不同方法在鹿类动物分子识别中的有效性。
1 材料和方法
1.1 样本收集
动物样本分为粪便样本和皮毛样本。以河麂(Hydropotesinermis,朝鲜亚种5个)、西伯利亚狍(Capreoluspygargus,7个)、梅花鹿(Cervusnippon,3个)、马鹿(Cervuselaphus,7个)、驯鹿(Rangifertarandus,1个)、麋鹿(Elaphurusdavidianus,3个)、黇鹿(Damadama,3个)和小麂(Muntiacusreevesi,1个)共8种鹿科动物30个样本作为研究对象。借助全国第二次陆生野生动物资源调查工作,在辽宁东部桓仁县和宽甸县采集朝鲜河麂粪便样本3份,其余2份为皮毛样本,采自老秃顶子国家级自然保护区标本馆馆藏河麂标本。其余动物粪便样本采自辽宁省辽阳县特种动物养殖场。粪便样本采集用一次性手套,装入塑料自封袋中置于液氮罐中冷冻带回,于-25℃冰箱中冷藏备用。
在GenBank和BOLD下载43条COI参考序列,除包括上述8个物种外,还有欧洲狍(Capreoluscapreolus)、黑麂(Muntiacuscrinifrons)、赤麂(Muntiacusmuntjak)3种鹿科动物以及马麝(Moschuschrysogaster)和林麝(Moschusberezovskii)2个麝科(Moschidae)物种在系统树构建中作为外群。
1.2 基因组DNA的提取、PCR扩增及测序
基因组提取试剂盒分别购自上海生工及QIAGEN、TIANGEN、AXYGEN公司,TaqDNA聚合酶购自大连宝生物工程公司,引物由北京华大基因公司合成。毛皮样本利用TIANGEN、AXYGEN试剂盒提取基因组,粪便样本用上海生工生产的磁珠法土壤试剂盒和QIAGEN粪便试剂盒提取基因组,-20℃条件下保存备用。
COI基因片段扩增采用一对通用引物(COI-F:5’-TTCATTAACCGCTGATTATTTTCAAC-3’;COI-R:5’-CACGATATGAGAAATTATACCAAACC-3’)和简并引物(DCIF15:5’-CGCAGGRGCTTCAGTAGAC-3’;DCIR12:5’-TRCCTCCRTGRAGTGTTGCT-3’)完成。PCR反应体积为25μl,其中Premix TaqTM(Ex TaqTM Version 2.0)混合液12.5μl,上下游引物各0.5μl、BSA 2.5μl、DNA模板2μl、超纯水7μl。PCR反应条件:95℃预变性7min,95℃变性45s,54℃退火45s,72℃延伸1min,36个循环。最后72℃延伸7min。每次PCR设立不含模板DNA的空白对照。在ABI2720 PCR仪上进行扩增,扩增产物经1%琼脂糖凝胶电泳检测后送北京华大基因有限公司测序。
1.3 DNA条形码序列分析
所测得COI序列经Blast搜索验证其可靠性,用MEGA 5软件[28]进行序列片段的拼接组装和多重比对,并将序列翻译成氨基酸以检验是否出现终止密码。把验证比对后的43条参考序列(reference)和30条查询序列(query)用于11种算法进行分析:条形码空隙探查法(ABGD)、邻接树法(NJ)、条形码逻辑公式法(BLOG)、最近邻法(1-NN)、决策树法(J48)、规则算法(Jrip)、随机森林法(RF)、支持向量机法(SVM)、反向传播算法(BP-based)、模糊集合算法(fuzzy set-based)和贝叶斯法(Bayesian-based)。
(1)NJ树:用MEGA 5软件构建基于K2P模型的NJ树,并进行1000次重复抽样的自展值检验。(2)ABGD:用ABGD软件估计分子可操作分类单元[29]。将查询序列以及由参考序列和查询序列组成的全部序列(reference+query)分别提交给在线软件(http://wwwabi.snv.jussieu.fr/public/abgd/abgdweb.html),选择K2P模型计算遗传距离,其余参数使用默认值。基于遗传距离对样本进行划分,将划分在同一组的样本认定为1个物种。(3)BLOG:将参考序列和查询序列按照文件格式要求分别输入BLOG 2.0软件[15],SCTYPE参数设为2,其余参数为缺省值。(4)1-NN、J48、Jrip、RF和SVM等5种分析方法在Weka 3.8.0软件[30]上运行。用Weka软件包中的fasta2weka程序将参考序列文件和查询序列文件分别转换为ARFF格式,后在Weka软件的Explorer模块下将参考序列文件输入程序,选择1-NN、J48、Jrip、RF和SVM等5种分类器分别对查询序列进行分析。(5)BP-based、Fuzzy set-based和Bayesian-based等3种分析方法用基于R的BarcodingR软件[31]进行。在ape程序包[32]下读入序列数据集并对参考序列与查询序列分布进行设置,用“bpNewTraining”“fuzzyId”和“Bayesian”命令进行相应分析。为评估种内以及种间遗传距离边界值,用BarcodingR软件进行了条形码空隙分析。
2 结果
30个样本中的鹿类动物线粒体COI基因部分序列经比对分析得到长度为700bp的片段。
2.1 NJ树
NJ树显示种间序列分歧明显大于种内分歧(图1),与目前鹿科动物分类系统一致。但其中马鹿样本被聚类为2个枝,表明马鹿种内COI基因存在较高的变异。
2.2 ABGD
以0.001~0.100的先验值P区间对查询序列30个样本进行划分,显示初始划分(initial partition)和递归划分(recursive partition)两种情况(图2)。初始划分较为稳定,30个样本均被分成7个组,而递归划分出现过度划分的情况。初始划分可操作分类单元数目除了将马鹿与梅花鹿合并为1个外,其余样本与采样物种一一对应,物种划分准确率87.5%。查询序列样本正确识别率90%。
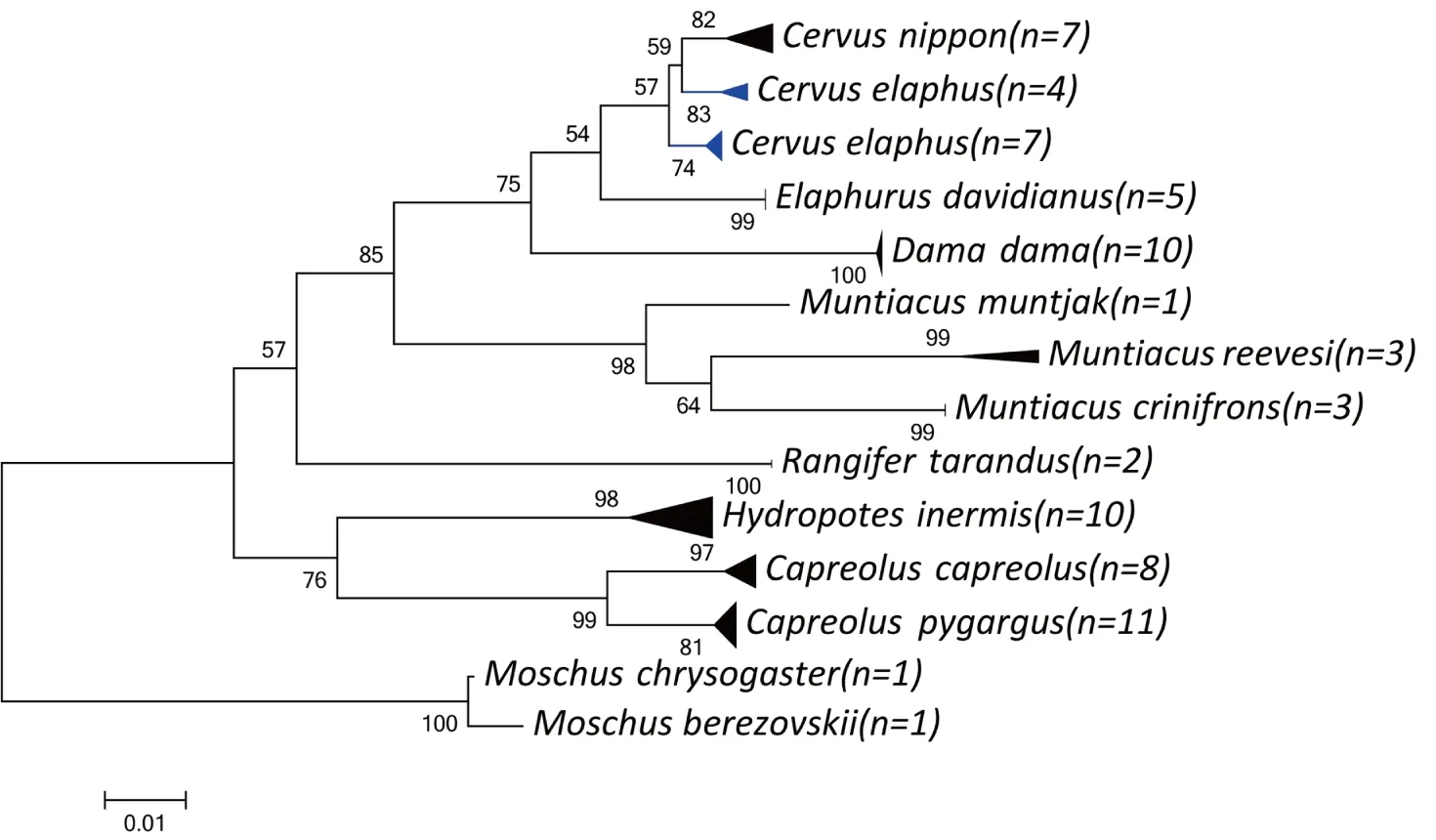
图1 30个样本13个物种的NJ树
2.3 BLOG
BLOG可成功识别所有参考序列特征碱基(表1)。对于查询序列BLOG成功识别21个样本,正确识别率为70%。未能识别序列9条,占查询序列的30%。未能识别序列来自马鹿5条、西伯利亚狍2条、河麂2条。

表1 基于43条COI参考序列的特征碱基诊断结果
2.4 1-NN
优化选择1-NN分类器模型对查询序列进行分类评估,成功识别分类26个样本,正确辨识率86.67%。3个黇鹿序列分类错误,被识别为马鹿,1个小麂样本被识别为梅花鹿,误报率为13.33%。
2.5 J48
基于决策树的J48算法正确识别查询序列中18个样本,占查询序列样本总数的60%。5个西伯利亚狍样本、1个黇鹿样本、3个麋鹿样本、2个河麂样本和1个驯鹿样本未能正确识别,占比40%。
2.6 Jrip
Jrip方法设定了8项规则,正确识别分类10个查询序列样本,包括2个西伯利亚狍样本、3个梅花鹿样本、2个黇鹿样本和3个河麂样本,正确识别率为33.33%。
2.7 RF
RF计算采用100次迭代抽样分析,正确识别了所有查询序列样本,正确辨识率100%。
2.8 SVM
SVM采用Linear Kernel模型,正确识别分类30个查询序列样本,正确识别率100%。
2.9 BP-based
采用人工智能的BP-based算法正确识别26个查询序列样本,正确识别率86.67%。识别错误的为3个黇鹿样本和1个小麂样本。
2.10 Fuzzy set-based
模糊数据集算法正确识别了3个河麂样本、3个梅花鹿样本和2个马鹿样本共8条查询序列,正确识别率26.67%。
2.11 Bayesian-based
贝叶斯算法的识别率很低,只有3个河麂和2个马鹿的查询序列样本被成功分类,仅占全部查询序列的16.67%。
3 讨论
本研究以8种鹿科动物为例,比较DNA条形码不同分析方法在该类动物分类中的解析能力和准确性。结果显示,11种分析方法在鹿类动物分类中的解析能力和准确性存在较大差异。NJ树分析除马鹿外,其它各物种的样本都能够独立聚类为一枝,而马鹿样本被分割为2个亚枝。马鹿与梅花鹿亲缘关系密切,属于近期分离物种,对二者的分子识别鉴定存在一定的复杂性和难度,这与Cai等(2015)[33]研究结果一致。其余10种可量化准确率的分析结果见图3,RF和SVM方法正确分类了全部30个查询序列样本,正确率达到100%,且二者间无显著差异(p>0.05)。ABGD、1-NN和BP-based 3种方法也有较强的解析能力,正确率达到或接近90%。而其他5种方法对鹿科动物DNA条形码的鉴别能力较弱,正确识别率小于(或等于)70%。RF和SVM法与其他方法鉴别结果存在显著差异(p<0.001)。

图3 可量化准确率的10种DNA条形码分析方法效果比较
DNA条形码分析方法不同,其机理和运算手段存在差异。SVM算法是具有较强识别力的分类器,它能够转化多维向量中的参考数据对象并定义对象中的分类超平面边界,以此作为不同分类标准。来自于查询序列的新对象按照这个分类超平面被评估分类。该方法最为重要的特性之一是它能够通过线性核函数进行有效的输入空间非线性转换,以实现高分类准确性[25,34-35]。在DNA条形码分类运算上,RF方法从参考序列中无重复地抽取多态位点作为子集建立大批量分类树,再用标记物种的多数一致树将查询序列样本分类到物种。上述2种方法实现了对检测样本的正确鉴别,显示其对鹿类动物分子识别的适用性。
ABGD自动探查DNA条形码序列对之间遗传距离分布的空隙,并找到空隙位置的距离值[29],将导入的查询序列分组到几个假定物种中。该方法对于有重叠分布的数据也能够有效分配,但要求有适当的最大种内分歧先验值,这个值一般在1%~3%之间,本研究为3%。少数情况下ABGD会出现多重物种臆测,即一个物种被分成2个(或相反),如递归划分的过度划分和马鹿与梅花鹿的并组现象。当数据中不存在条形码空隙时ABGD就不能作出基本的分类单元划分。
BLOG诊断方法以DNA条形码关键核苷酸位点作为物种简单特征,形成逻辑规则并以此作为分类依据诊断查询序列样本的物种归属。如麋鹿的识别:如果348位点是C,那么这个序列样本就是麋鹿。BLOG诊断方法对查询序列样本有较高的正确识别率[36]。
1-NN方法把参考序列与查询序列按照K2P距离远近排序,将查询序列中最近邻样本归类到所属物种,如果有2个以上查询序列样本具有相同距离时则将它们归为一组同级别类群[14]。Jrip算法通过对数据集进行重复增量修剪来构建一套初始规则,再利用这套优化规则对查询序列样本逐一分类,直到对所有样本的全覆盖。J48分类法按照信息增益为树的每个节点寻找最佳分裂点和最优特征,但所谓决策树并非终结的顶点,其结构简单,仅代表着1个或几个检验属性。由于参考序列变异性会产生不同属性特征,因而决策树有不稳定的弱点。
BP-based算法借鉴最新人工智能技术,通过定义神经网络对参考序列网络进行验证,最后利用经培训的网络对查询序列样本进行识别分类。模糊集合是经典数据集概念的扩展,不同于二进制评估经典理论,模糊集理论对数据中各元素逐级评估,对于数据不完整或不精确的生物信息分析有很好的应用价值。该方法通过定义物种从属函数,搜索查询序列样本最邻近的潜在物种。
DNA条形码技术的目的是利用小DNA片段数据来实现对查询序列样本的物种分类[37],这个小片段必须有高信息含量。由于不同物种间可能会共享许多多态位点(它们在祖先物种中或许是多态的),这些多态位点的固定以及突变在各自物种中的特异体现都需要相当长的时间,因而对现生的近期分离物种用DNA条形码进行物种鉴定存在一定困难。本研究选取的11种分析方法在近期与远古分离物种识别能力上就存在差异,总体上看对近期分离物种的识别能力低于远古物种。另外,由于不完全的世系排序或低突变率以及缺少条形码特征位点[37],DNA条形码的识别能力会受到很大限制。这些问题可以通过增加取样数量、增加测序长度或增加基因位点(如核基因)等手段,适当增加信息含量加以解决。
4 结论
本研究证明,基于COI基因的DNA条形码能够有效和准确识别大多数鹿科动物。从比较结果看,没有普遍适用的方法,RF和SVM法识别正确率最高。对识别检测性能影响最大的是DNA分子的多态性程度,通过增加DNA条形码信息含量或选择对分类样本最为适用的分析方法,提高多数分析方法的预测性能。
[1]Hebert PDN,Cywinska A,Ball SL,et al.Biological identifications through DNA barcodes[J].Proc. R.Soc.Lond. B,2003,270(1512):313-321.
[2]Hebert PDN,Ratnasingham S,deWaard JR.Barcoding animal life:cytochrome c oxidase subunit 1 divergences among closely related species[J].Proc.R.Soc. Lond. B(Suppl.)2003,270(Suppl_1):S96-S99.
[3]Ratnasingham S,Hebert PDN.BOLD:The barcode of life data system(www.barcodinglife.org)[J].Molecular Ecology Notes,2007,7(3):355-364.
[4]Dellicour S,Flot JF.Delimiting species-poor data sets using single molecular markers: A study of barcode gaps, haplowebs and GMYC[J].Systematic Biology,2015,64(6):900-908.
[5]Meier R,Shiyang K,Vaidya G,et al.DNA barcoding and taxonomy in Diptera:a tale of high intraspecific variability and low identification success[J].Syst Biol,2006,55(5):715-728.
[6]Schmidt S,Schmid-Egger C,Morinière J,et al.DNA barcoding largely supports 250 years of classical taxonomy: identifications for Central European bees (Hymenoptera, Apoidea partim)[J].Molecular Ecology Resources,2015, 15(4):985-1000.
[7]Li J,Zheng X,Cai Y,et al.DNA barcoding of Murinae (Rodentia: Muridae) and Arvicolinae(Rodentia:Cricetidae) distributed in China[J].Molecular Ecology Resources,2014,15(1):153-167.
[8]Clare EL,Lim BK,Engstrom MD,et al.DNA barcoding of Neotropical bats:species identification and discovery within Guyana[J].Molecular Ecology Notes,2007,7(2):184-190.
[9]何锴,王文智,李权,等.DNA 条形码技术在小型兽类鉴定中的探索:以甘肃莲花山为例[J].生物多样性,2013,21(2): 197-205.
[10]马英,李海龙,鲁亮,等.DNA 条形码技术在青海海东地区小型兽类鉴定中的应用[J].生物多样性,2012,20(2):193-198.
[11]Saitou N,Nei M.The neighbour-joining method: a new method for reconstructing phylogenetic trees[J].Mol Biol Evol Appl,1987,4(4):406-425.
[12]Farris JS.Estimating phylogenetic trees from distance matrices[J].Am Nat,1972,106(951):645-668.
[13]Munch K,Boomsma W,Huelsenbeck JP,et al.Statistical assignment of DNA sequences using Bayesian phylogenetics [J].Systematic Biology,2008,57(5):750-757.
[14]Austerlitz F,David O,Schaeffer B,et al.DNA barcode analysis: a comparison of phylogenetic and statistical classification methods[J].BMC Bioinformatics,2009,10(14):S10.
[15]Weitschek E,Velzen R,Felici G,et al.BLOG 2.0: a software system for character-based species classification with DNA barcode sequences.What it does,how to use it[J].Molecular Ecology Resources,2013,13(6):1043-1046.
[16]Dasgupta B,Konwar KM,Ndoiu II,et al.DNA-BAR:distinguisher selection for DNA barcoding[J].Bioinformatics, 2005,21(16):3424-3426.
[17]Little DP.DNA barcode sequence identification incorporating taxonomic hierarchy and within taxon variability [J].PLoS ONE,2011,6(8):e20552.
[18]Little DP.BRONX2:Barcode recognition obtained with nucleotide eXposés [R].2012.
[19]Nielsen R,Matz M.Statistical approaches for DNA barcoding[J].Systematic Biology,2006,55(1):162-169.
[20]Matz MV,Nielsen R.A likelihood ratio test for species membership based on DNA sequence data[J].Phil.Trans.R.Soc.B,2005,360(1462):1969-1974.
[21]Abdo Z,Golding GB.A Step toward barcoding life:a model-based,decision-theoretic method to assign genes to preexisting species groups[J].Systematic Biology,2007,56(1):44.
[22]Zhang AB.Inferring species membership using DNA sequences with back-propagation neural networks[J].Systematic Biology,2008,57(2):202-215.
[23]Zhang AB,Feng J,Ward RD,et al.A new method for species identification via protein-coding and non-coding DNA barcodes by combining machine learning with bioinformatic methods[J].PLoS ONE,2012,7(2):e30986.
[24]Zhang AB,Muster C,Liang HB,et al.A fuzzy-set-theory-based approach to analyse species membership in DNA barcoding[J].Molecular Ecology,2012,21(8):1848-1863.
[25]Weitschek E,Fiscon G,Felici G.Supervised DNA barcodes species classification:analysis,comparisons and results[J].BioData Mining,2014,7(1):4.
[26]王应祥.中国哺乳动物种和亚种分类明录与分布大全[M].北京:中国林业出版社,2003.
[27]蒋志刚,江建平,王跃招,等.中国脊椎动物红色名录[J].生物多样性,2016,24(5):500-551.
[28]Tamura K,Peterson D,Peterson N,et al.MEGA5: Molecular evolutionary genetics analysis using maximum likelihood,evolutionary distance, and maximum parsimony methods[J].Molecular Biology and Evolution,2011, 28(10):2731-2739.
[29]Puillandre N,Lambert A,Brouillet S,et al.ABGD,Automatic barcode gap discovery for primary species delimitation[J].Molecular Ecology,2012,21(8):1864-1877.
[30]Hall M,Frank E,Holmes G,et al.The WEKA data mining software: an update[J].SIGKDD Explorations, 2009,11(1):10-18.
[31]Zhang AB,Hao MD,Yang CQ,et al.Barcoding R:an integrated R package for species identification using DNA barcodes[J].Methods in Ecology and Evolution,2016,DOI:10.1111/2041-210X.12682.
[32]Paradis E,Claude J,Strimmer K.APE:Analyses of phylogenetics and evolution in R language[J]. Bioinformatics, 2004,20(2):289-290.
[33]Cai Y,Zhang L,Wang Y,et al.Identification of deer species(Cervidae,Cetartiodactyla) in China using mitochondrial cytochrome c oxidase subunit I(mtDNA COI)[J].Mitochondrial DNA,2015:1-4.
[34]Fischetti M.Fast training of support vector machines with gaussian kernel[J].Discrete Optimization,2015(22):183-194.
[35]Kuksa P,Pavlovic V.Fast kernel methods for SVM sequence classifiers[J].Lecture Notes in Computer Science,2007,4645:228-239.
[36]Van Velzen R,Weitschek E,Felici G,et al.DNA barcoding of recently diverged species:relative performance of matching methods[J].PLoS ONE,2012,7(1):e30490.
[37]Meusnier I,Singer G,Landry JF,et al.A universal DNA mini-barcode for biodiversity analysis [J].BMC Genomics, 2008,9(1):214.
DNA Barcode Analysis: A Comparison of Performance Between Different Classification Methods in Deer Species
MA Wei-wei, LIU Yi-ming, DONG Bing-jun, YANG Bao-tian
(College of Life Science, Shenyang Normal University, Shenyang Liaoning 110034,China)
DNA barcoding aims to assign individuals to given species according to their sequence at a small locus, generally part of the COI gene. In this context, we examined 11 assignation methods in 8 deer species and investigated the ability of each method to correctly classify 30 query sequences. The results indicated that both of random forest and support vector machines were found to be the most reliable with respect to the data sets from deer. No method was found to be the best in all cases. The element most influencing the performance of the various methods was molecular diversity of the data. All of addition of genetically independent loci, lengthening sequences and increasing the sample size improved the predictive performance of most methods. The study implies that the quality of analyses was enhanced by choosing a method best-adapted to the sample.
COI;DNA barcoding;Cervidae;species identification
2016-12-20
辽宁省自然科学基金指导计划项目“东北林蛙个体分子鉴别与群体遗传学研究”(201602677)。
马巍威(1990- ),男,硕士研究生,从事分子生态学研究。
杨宝田(1963- ),男,副教授,硕士生导师,博士,从事野生动物保护及分子生态学研究。
Q959.5+3
A
2095-7602(2017)04-0054-07
猜你喜欢
少年文艺·开心阅读作文(2021年8期)2021-09-05
计算机工程(2020年3期)2020-03-19
中国听力语言康复科学杂志(2019年3期)2019-06-24
小学科学(学生版)(2019年5期)2019-05-21
少儿美术(快乐历史地理)(2019年11期)2019-04-20
中国交通信息化(2018年3期)2018-06-13
小哥白尼(野生动物)(2018年1期)2018-05-26
小学生导刊(2017年13期)2017-06-15
中国交通信息化(2016年2期)2016-06-06
新疆大学学报(自然科学版)(中英文)(2016年4期)2016-05-16
