
CRISPR/Cas9介导的高产谷胱甘肽原养型酵母工程菌的构建
2017-04-16 05:22:48周文龙唐亮成凯刘忞之杨燕王伟
生物工程学报 2017年12期
周文龙,唐亮,成凯,刘忞之,杨燕,王伟
中国医学科学院 北京协和医学院药物研究所 天然药物活性物质与功能国家重点实验室/国家卫生和计划生育委员会天然药物生物合成重点实验室,北京 100050
谷胱甘肽 (Glutathione,GSH) 是细胞内含量最多的非蛋白质类巯基化合物,由谷氨酸、半胱氨酸和甘氨酸经过酶催化反应合成[1]。GSH因其具有游离的巯基而具有很多生物活性,如抗氧化[2]、脱毒[3]、免疫调节[4]等。体内GSH缺失与多种疾病有关,如夸休可尔症、癫痫、阿尔茨海默病等[5]。因此,GSH被广泛应用于药品、食品、化妆品等行业。目前,GSH在国内主要用于药物领域,作为肝病和癌症治疗后修复损伤的辅助用药,在国外则以食品添加剂及保健品为主。GSH市场需求逐年增加,有人预测至2019年,全球GSH需求量将达200–300 t[6]。
目前,GSH生产的主要方法是酵母发酵法。由于野生型酿酒酵母Saccharomyces cerevisiae和产朊假丝酵母Candida utilis细胞中 GSH含量高,因此,它们是GSH发酵生产的重要菌株。提高GSH产量的方法有提高细胞内GSH含量、提高细胞生物量[7]、缩短菌株培养周期等。提高细胞内GSH含量主要通过遗传育种完成,目前主要有经典筛选 (Classical selection)、遗传工程育种等方法。提高细胞生物量主要通过优化发酵培养基、优化发酵工艺条件等途径来实现[7]。菌株培养周期则与菌株所处环境和菌株自身遗传背景密切相关。
Murata等分别在1981年和1983年克隆并测序了编码GSH生物合成的两个酶即γ-谷氨酰半胱氨酸合成酶 (GSHⅠ) 和GSH合成酶 (GSHⅡ)的基因gshA和gshB[1],之后科学家们又在一些革兰氏阳性菌中发现了能够独立完成 GSH生物合成的双功能酶GshF (由gshF编码)[8-9]。随着参与GSH代谢酶的编码基因不断被分离和鉴定,科学家们利用多种遗传工程策略提高模式微生物细胞内GSH的产量,在这些遗传操作过程中需要用到抗性或者营养缺陷型等遗传标记筛选阳性克隆。其中营养缺陷型菌株为初级代谢途径阻断变异型,仅能在一些特定的培养基中正常生长。
近年来,串联间隔短回文重复序列(Clustered regularly interspaced short palindromic repeats,CRISPR) 及 RNA 靶向内切酶 Cas9(CRISPR-Cas9) 介导的基因组编辑技术发展迅速并得以广泛应用。该技术通过一段短的引导RNA (Guide RNA,gRNA) 识别特定的DNA序列,通过改变gRNA序列即可使Cas9定位到新的DNA序列。目前该技术已广泛应用于多种生物如人、小鼠、斑马鱼等的基因编辑[10]。
本研究旨在通过在酿酒酵母中建立 CRISPR/Cas9介导的基因组编辑技术,回复突变工程菌株W303-1b/FGP为原养型菌株,使其能够自主合成生命活动中必需的小分子化合物,能在简单的无机盐培养基中生长,方便大规模培养。
1 材料与方法
1.1 菌株与培养基
大肠杆菌Escherichia coliTrans1-T1 (TransGen公司) 用于重组DNA的扩增与构建。酿酒酵母工程菌株 W303-1b/FGP (MATα ade2-1 leu 2-3,112 his3-11,15 ura3-1 trp1-1) (表 1)[11]为出发菌株。
YPD培养基:酵母提取物10 g/L,蛋白胨20 g/L,葡萄糖20 g/L。
1.2 质粒构建与酵母转化
1.2.1 Cas9表达载体的构建
以质粒pGADT7 (Clontech) 为基础构建含有编码Cas9基因的表达载体,首先用以酵母基因组DNA为模板和引物PGK1_Sph/PGK1_Nde扩增3-磷酸甘油酸激酶基因启动子 (PGK1),此后用限制性内切酶位点SphⅠ和NdeⅠ定向亚克隆到质粒 pGADT7中,即得到 pGAD-PGK1。由于编码 Cas9的基因较长,分 2步进行亚克隆到质粒pGAD-PGK1中。先用质粒模板pX260 (Addgene)和引物X260_1/X260_2扩增2.2 kb片段,用限制性内切酶NdeⅠ和EcoRⅠ亚克隆到 pGADPGK1;然后再用X260_2/X260_4扩增1.8 kb片段,用限制性内切酶SacⅠ和XhoⅠ亚克隆,即得到含有Cas9编码框的质粒pGAD-Cas9。为消除质粒 pGAD-Cas9的筛选标记leu2,利用引物GADT7_H3/GADT7_Nhe和质粒pGAD-Cas9为模板扩增ADH1终止子,再用相同的内切酶Hind Ⅲ和NheⅠ置换相应的 DNA区段,消除长度为1.9 kb的筛选标记Leu2基因,并保留原始终止子ADH1序列不变,最后利用引物Bla_Nhe/Bla_Not和pPIC6 (Invitrogen公司) 质粒为模板扩增含有杀稻瘟菌素 (Blasticidin) 抗性筛选标记的基因片段,用限制性内切酶NheⅠ和NotⅠ亚克隆到消除筛选标记Leu2的pGAD-Cas9质粒中,即获得含有抗性筛选标记 (Bla) 并组成型表达Cas9的表达载体pGAD-Cas9-Bla。
1.2.2 gRNA转录表达框的构建
以DiCarlo等报道的方法[13],利用引物 (表2)SNR_G1到SNR_G7和Ade_G1经过连续PCR的方法获得含有酿酒酵母 snoRNA SNR52启动子和一个靶向ade2的gRNA序列的295 bp DNA片段;同时以引物Ade_G2和SNR_G8到SNR_G9经连续PCR方法获得含有gRNA结构片段和酿酒酵母SUP4终止子3′区的120 bp DNA片段;上述2个片段进行DNA变性和退火补平反应,再利用引物SNR_G1和SNR_G9进行PCR扩增,即得到能转录表达靶向ade2的gRNA结构片段(gRNA (Ade))。最后将该片段进行“TA”克隆到载体EZ-T (北京康润科技有限公司),得到质粒EZ-gRNA-Ade。其余靶向酿酒酵母ura3、leu2、trp1、his3基因的 gRNA表达框分别用引物Ura_G1、Leu_G1、Trp_G1、His_G1 与 M13+和引物 Ura_G2、Leu_G2、Trp_G2、His_G2 与 M13–为引物配对,以 EZ-gRNA-Ade为模板,进行PCR反应,然后以上轮 PCR扩增得到的 2个DNA片段进行补平反应,再以补平后的 DNA片段为模板,再利用引物 SNR_G1和 SNR_G9进行 PCR扩增,即得到能转录表达 gRNA的DNA 结构片段,即 gRNA (Ura3)、gRNA (Leu2)、gRNA (Trp1)、gRNA (His3)。
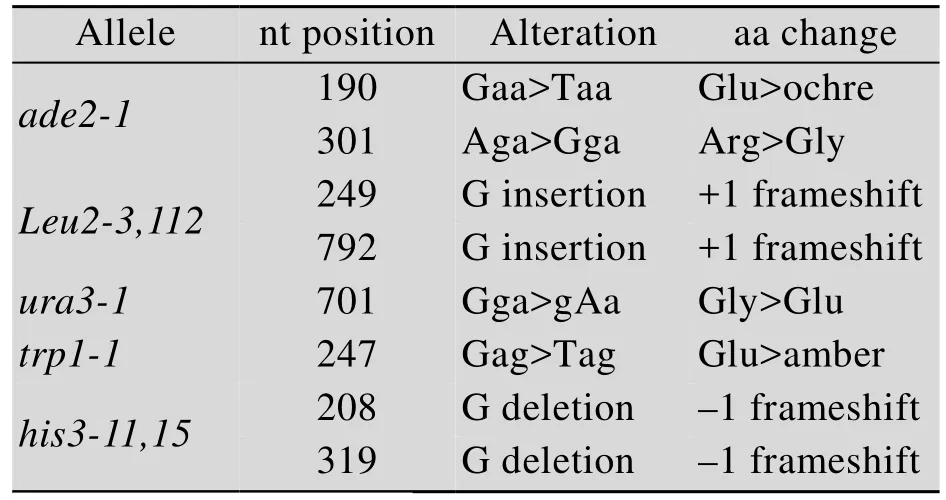
表1 工程菌株W303-1b/FGP基因型Table 1 W303-1b/FGP genotype

表2 本实验所使用PCR扩增引物Table 2 PCR primers used in this study
1.2.3 目的基因 DNA片段的扩增、回复突变及阳性克隆的鉴定
分别以野生型酿酒酵母S288C基因组DNA或含有目的基因的质粒为模板,以Ade1/Ade2、Ura1/Ura2、Leu1/Leu2、Trp1/Trp2、His1/His2为引物对,利用PCR扩增分别编码ade2、ura3、1eu2(pGADT7)、trp1(pGBKT7) 和his3的目的基因片段,电泳定量观察。回复突变实验分为3个阶段进行:首先利用醋酸锂法[14]将表达Cas9的质粒载体pGAD-Cas9-Bla转化W303-1b/FGP,用75 μg/mL杀稻瘟菌素进行筛选;用转录表达gRNA (Ade2)、gRNA (Ura3) 的结构DNA片段各1 μg和用于回复突变ade2、ura3所对应的基因片段各5 μg共转化表达Cas9的感受态细胞,用含有杀稻瘟菌素和不添加Ade和Ura 合成培养基进行筛选,获得的阳性克隆即为回复突变了ade2、ura3这2个基因的阳性克隆;然后在此基础上进行第2次回复突变1eu2、trp1位点;最后回复突变his3位点,即获得了回复突变5个营养缺陷筛选标记 (ade2、ura3、1eu2、trp1和his3) 的原养型工程菌W303-1b/FGPPT。
提取原养型工程菌W303-1b/FGPPT的基因组DNA,分别利用引物对Ade1/Ade2、URA1/URA2、Leu1/Leu2、Trp1/Trp2、His1/His2进行 PCR扩增,然后纯化PCR扩增的DNA片段;为确保回复突变位点测序结果的准确性,分别合成测序引物 Ade3、Ura3、Leu3、Leu4、Trp3、His3进行测序验证,使得回复突变位于每个测序引物所测得的图谱最准确的150–600 bp之间。
1.3 酿酒酵母工程菌株摇瓶培养
挑取回复突变的工程菌株 3个阳性克隆于20 mL YPD液体培养基中,30 ℃、250 r/mim培养 18–24 h作为种子液,转接适当体积种子至50 mL新鲜的YPD培养基中,使得培养液初始OD600为0.2,30 ℃、220 r/min条件下培养。
1.4 分析方法
1.4.1 细胞干重的测量
收集 5 mL菌液,灭菌双蒸水漂洗 2次,100 ℃下烘干24 h。
1.4.2 浊度测量
将培养一段时间的培养液稀释至适当浓度,利用紫外分光光度计测定600 nm下的吸光度值 (OD600)。
1.4.3 细胞内GSH的萃取
利用 40%乙醇萃取的方法萃取酿酒酵母细胞内的GSH[11]。
1.4.4 细胞内GSH的产量
利用 4-(氨磺酰)-7-氟 2,1,3-苯并恶二唑 (ABDF) 衍生化法测定萃取液中GSH的浓度[15]。分析柱为YMC-Pack ODS-A 250 mm×4.6 mm。D.S-5 µm,30 nm。梯度洗脱条件为:0–14 min,8%B 相;15–18 min,8%–90% B 相;19–22 min,90% B 相;23–26 min,90%–8% B 相;27–30 min,8% B相;流速1 mL/min,检测波长为390 nm,以GSH浓度与峰面积形成的曲线为标准曲线。
1.5 原养型菌株GSH产量稳定性分析
挑取3个原养型单克隆于10 mL液体WMVIII培养基[16]中,30 ℃、250 r/mim条件下培养至OD600为15–30,转接至新鲜的10 mL液体WMVIII培养基中,并使初始OD600=0.5,再培养至OD600为15–30,重新转接,共转接20次,每转接5次测定菌株GSH产量。
2 结果
2.1 CRISPR/Cas9系统的建立
目前文献报道用于酿酒酵母的 CRISPR/Cas9基因编辑系统都是使用含有营养缺陷型筛选标记如ura3的质粒载体[13,17],与本研究所要获得的原养型工程菌株相矛盾,于是以质粒pGADT7为骨架,构建了含有抗性筛选标记Bla并使用转录活性较弱的启动子PGK1表达Cas9的质粒载体 (图1A);gRNA的转录直接通过PCR方法构建融合表达框。
2.2 原养型工程菌株的构建
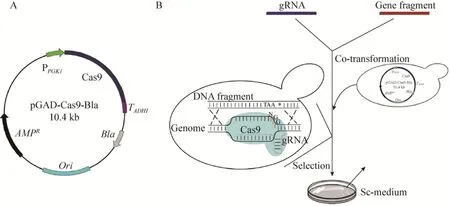
图1 酿酒酵母原养型菌株构建示意图 (A:Cas9表达载体pGAD-Cas9-Bla示意图;B:CRISPR/Cas9介导的酿酒酵母营养缺陷型的回复突变)Fig. 1 Schematic diagram of the reversion of the auxotrophic strain. (A) Schematic diagram of pGAD-Cas9-Bla. (B)The reversion of the auxotrophic strain mediated by CRISPR/Cas9.
将质粒pGAD-Cas9-Bla转化进入酿酒酵母工程菌株W303-1b/FGP中,将阳性克隆命名为W303-1b/FGP/Cas9。然后,分 3次进行原养型回复突变,依次是ade2和ura3、leu2和trp1、his3基因。其操作过程主要是把野生型的目的基因片段和转录合成靶向相应基因的 gRNA结构 DNA片段按 5∶1的比例共转化进入菌株W303-1b/FGP/Cas9中,其中利用CRISPR/Cas9基因编辑技术同时进行多个基因的编辑操作已得到实验证实[17-18];利用不添加相应的营养因子的SC合成培养基进行阳性克隆筛选,最终获得回复为原养型的工程菌株W303-1b/FGPPT,基本过程见图1B。在正向选择压力下,获得阳性克隆率为 100%,利用 PCR方法扩增阳性克隆的靶基因DNA片段并测序验证 (图2)。
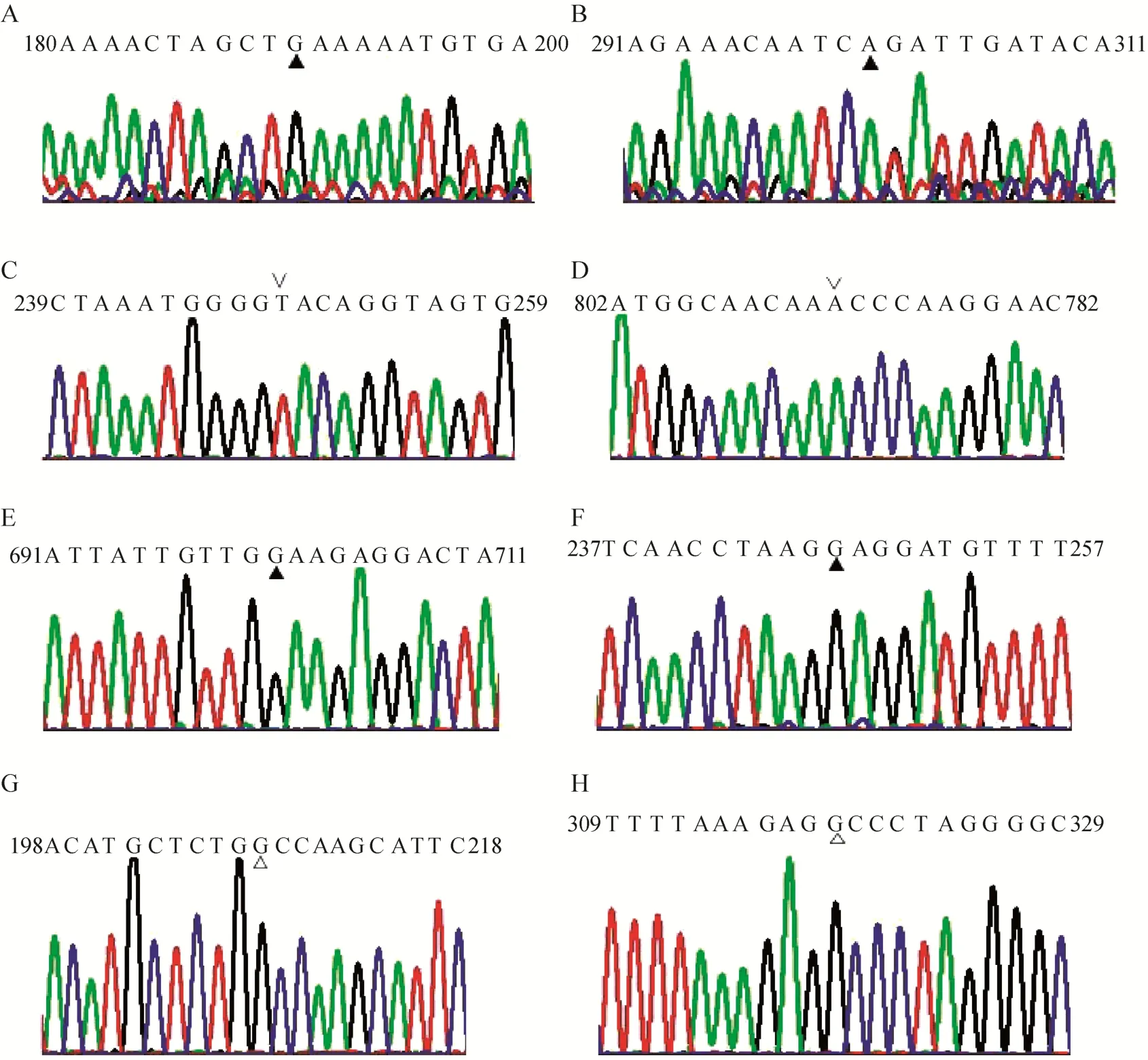
图2 酿酒酵母原养型菌株回复突变位点的测序结果 (▲:回复的突变位点;∨:回复的插入突变位点;△:缺失突变位点. A:ade2-1 T190G,赭石突变位点的回复;B:ade2-1 G301A,Gly>Arg的回复突变;C:Leu2-3,112, T249插入的G缺失回复;D:Leu2-3,112, A792插入的G缺失回复;E:ura3-1 A701G, Glu>Gly的回复突变;F:trp1-1 T190G,琥珀突变位点的回复;G:his3-11,15 G208的缺失回复;H:his3-11,15 G319的缺失回复)Fig. 2 The sequencing results of reverse allelic sites. ▲: reverse mutation. ∨: reverse insertion; △: reverse deletion.(A) ade2-1 T190G, reverse ochre mutation. (B) ade2-1 G301A, Gly>Arg reverse mutation. (C) Leu2-3,112, G249 reverse an insertion. (D) Leu2-3,112, G792 reverse an insertion. (E) ura3-1 A701G, Glu>Gly reverse mutation. (F)trp1-1 T190G, reverse amber mutation. (G) his3-11,15 G208 reverse deletion. (H) his3-11,15 G319 reverse deletion.
2.3 营养缺陷型菌株与原养型菌株的培养周期
为观察营养缺陷型酿酒酵母工程菌株W303-1b/FGP与原养型工程菌株W303-1b/ FGPPTGSH产量及生物量的差别,将两种菌株同时在YPD培养基中培养,在不同时间内测定两种菌株的细胞内GSH产量及生物量。结果如图3所示。由图3A可知,营养缺陷型菌株W303-1b/FGP与原养型工程菌株W303-1b/FGPPT的GSH最大产量均为216 mg/L左右。由图3B可知,二者的最高生物量 (DCW) 为9.3 g/L。但是菌株W303-1b/FGP的生物量在培养 96 h时达到最大,而菌株W303-1b/FGPPT在培养48 h时即达到最大生物量。
2.4 原养型菌株的GSH产量稳定性
为考察工程菌株回复为原养型后GSH产量的稳定性,将菌株 W303-1b/FGPPT在无机盐培养基 WMVIII中进行连续培养,最初培养液OD600为 0.5,当OD600达到 15–30时转移至新鲜的培养基中进行培养,每转接 5次观察一下菌株最大生物量及GSH产量是否发生变化,共转接20次,结果如表3和图4所示,连续培养过程中菌株 GSH产量与连续培养前的比较采用 T检验,比较结果P>0.05,认为没有统计学差异。由表3和图4的结果可知,菌株W303-1b/FGPPT在 WMVIII培养基中连续培养 100代后,菌株GSH产量没有改变。
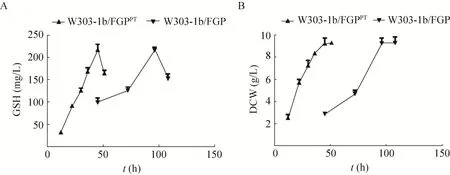
图3 YPD培养基中工程菌株W303-1b/FGPPT和W303-1b/FGP的GSH产量 (A) 及生物量 (B) 随时间的变化Fig. 3 GSH production (A) and cell growth (B) of W303-1b/FGPPT and W303-1b/FGP in YPD medium.

表3 原养型菌株连续培养100代GSH产量Table 3 GSH production of the prototrophic strain in 100 generations
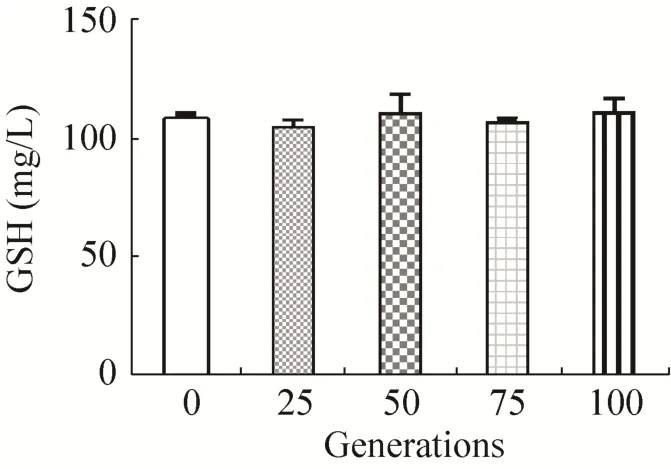
图4 工程菌株W303-1b/FGPPT合成GSH的稳定性Fig. 4 The stability of GSH production of W303-1b/FGPPT strain.
3 讨论
近年来,基因组编辑技术快速发展,其中CRISPR/Cas9技术因设计操作简便、编辑高效与通用性广等优点而备受关注[10],在酿酒酵母中也同样有文献报道[13,17-19]。但这些研究都使用了酵母细胞自身的营养缺陷型筛选标记如trp1、leu2、ura3等,并且还使用了双质粒表达系统或单个质粒同时表达Cas9和gRNA;实验操作过程复杂,花费时间长。尽管也有文献报道一步多基因编辑的成功操作,但也需要构建多个gRNA表达框串联的质粒表达载体[17-18]。本研究为简化实验操作,首次成功地直接利用 gRNA融合表达框转录表达目的gRNA,同时还探索研究了 2个基因同时进行基因编辑的可行性并在正向选择压力情况下获得成功,这为更多基因座位的编辑提供实验借鉴。
2013年酵母产品的全球市场已达到58亿美元,预计2019年将达到92亿美元[20],GSH就是其中一个具有广泛使用范围的产品。在大多数原核和真核细胞中,GSH是含量最丰富的非蛋白质类巯基化合物,浓度可达0.2–10 mmol/L[21]。细胞内还原型 GSH与氧化型谷胱甘肽 (GSSG)比例可达到30∶1–100∶1,形成氧化还原电势为E0=–240 mV,此电势对维持细胞内适宜的氧化还原状态非常重要,如清除细胞内多余的自由基和重金属等[22]。因此,GSH可以用于治疗GSH缺失的疾病以及化妆品行业。另外,含GSH丰富的酵母物质因其特殊的kokimi风味而被用于食品工业[23]。
目前生产 GSH所用的菌株主要为酿酒酵母和产朊假丝酵母,其中产朊假丝酵母中GSH最高产量可达 2.45 g/L[24]。利用基因工程改造的微生物也有可能得到GSH产量极高的菌株,例如在大肠杆菌Escherichia coliJM109 (pTrc99A-gshF)中,GSH产量可达11.30 g/L[25]。为提高GSH产量,科学家们做了一系列研究,其中两个主要的方法是提高生物量和细胞内GSH含量[7]。
研究组前期获得了组合表达3条GSH合成途径的酵母工程菌 W303-1b/FGP[11],但该工程菌株存在多个营养缺陷型基因,难以实现在无机盐培养基的发酵培养。为消除这些营养缺陷型基因导致的严重不足,利用CRISPR/Cas9介导的基因组编辑技术,将营养缺陷型酿酒酵母菌株W303-1b/FGP回复为原养型W303-1b/FGPPT后,在摇瓶水平下达到GSH产量最高所需的时间由96 h缩短为48 h,发酵周期大大缩短,单位时间GSH产量提高1倍。
与营养缺陷型菌株相比,原养型菌株一个很大的优点是可以自身合成Trp等必需小分子,这就使得原养型菌株可以在简单的、化学成分确定的无机盐培养基 (Chemical defined medium,CDM) 中正常生长,而不需要额外添加昂贵的氨基酸。利用CDM进行培养有从实验室摇瓶培养向规模化放大生产转化更快、过程再现强以及下游操作简单等优点[26]。
原养型菌株在 WMVIII培养基中连续培养100代GSH产量不发生明显变化,说明该菌株GSH生产能力稳定,上游基因改造不会在长时间培养过程中发生丢失,有利于菌株更深入的工程改造和放大培养。
[1] Murata K, Kimura A. Cloning of a gene responsible for the biosynthesis of glutathione inEscherichia coliB. Appl Environ Microbiol, 1982,44(6): 1444–1448.
[2] Jones DP. Redefining oxidative stress. Antioxid Redox Signal, 2006, 8(9/10): 1865–1879.
[3] Bock KW, Lilienblum W, Fischer G, et al. The role of conjugation reactions in detoxication. Arch Toxicol, 1987, 60(1/3): 22–29.
[4] Dröge W, Breitkreutz R. Glutathione and immune function. Proc Nutr Soc, 2000, 59(4): 595–600.
[5] Wu GY, Fang YZ, Yang S, et al. Glutathione metabolism and its implications for health. J Nutr,2004, 134(3): 489–492.
[6] Lorenz E, Schmacht M, Stahl U, et al. Enhanced incorporation yield of cysteine for glutathione overproduction by fed-batch fermentation ofSaccharomyces cerevisiae. J Biotechnol, 2015,216: 131–139.
[7] Li Y, Wei GY, Chen J, et al. Glutathione: a review on biotechnological production. Appl Microbiol Biotechnol, 2004, 66(3): 233–242.
[8] Gopal S, Borovok I, Ofer A, et al. A multidomain fusion protein inListeriamonocytogenescatalyzes the two primary activities for glutathione biosynthesis. J Bacteriol, 2005, 187(11): 3839–3847.
[9] Janowiak BE, Griffith OW. Glutathione synthesis inStreptococcus agalactiaeone protein accounts for γ-glutamylcysteine synthetase and glutathione synthetase activities. J Biol Chem, 2005, 280(12):11829–11839.
[10] Harrison MM, Jenkins BV, O’Connor-Giles KM,et al. A CRISPR view of development. Genes Dev,2014, 28(17): 1859–1872.
[11] Tang L, Wang WW, Zhou WL, et al. Threepathway combination for glutathione biosynthesis inSaccharomyces cerevisiae. Microb Cell Fact,2015, 14(1): 139.
[12] Ralser M, Kuhl H, Ralser M, et al. TheSaccharomyces cerevisiaeW303-K6001 crossplatform genome sequence: insights into ancestry and physiology of a laboratory mutt. Open Biol,2012, 2(8): 120093.
[13] DiCarlo JE, Norville JE, Mali P, et al. Genome engineering inSaccharomyces cerevisiaeusing CRISPR-Cas systems. Nucleic Acids Res, 2013,41(7): 4336–4343.
[14] Schiestl RH, Gietz RD. High efficiency transformation of intact yeast cells using single stranded nucleic acids as a carrier. Curr Genet,1989, 16(5/6): 339–346.
[15] Steele ML, Ooi L, Münch G. Development of a high-performance liquid chromatography method for the simultaneous quantitation of glutathione and related thiols. Anal Biochem, 2012, 429(1): 45–52.
[16] Patzschke A, Steiger MG, Holz C, et al. Enhanced glutathione production by evolutionary engineering ofSaccharomyces cerevisiaestrains. Biotechnol J,2015, 10(11): 1719–1726.
[17] Jakočiūnas T, Bonde I, Herrgård M, et al.Multiplex metabolic pathway engineering using CRISPR/Cas9 inSaccharomyces cerevisiae.Metab Eng, 2015, 28: 213–222.
[18] Bao ZH, Xiao H, Liang J, et al. Homologyintegrated CRISPR-Cas (HI-CRISPR) system for one-step multigene disruption inSaccharomyces cerevisiae. ACS Synth Biol, 2015, 4(5): 585–594.
[19] Xu K, Ren CH, Liu ZT, et al. Efficient genome engineering in eukaryotes using Cas9 fromStreptococcus thermophilus. Cell Mol Life Sci,2015, 72(2): 383–399.
[20] Marz U. Yeasts, Yeast Extracts, Autolysates and Related Products: the Global Market. BCC Research Report Code: CHM053B, Wellesley, MA:BCC Research, 2014.
[21] Anderson ME. Glutathione: an overview of biosynthesis and modulation. Chem Biol Interact,1998, 111–112: 1–14.
[22] Ayer A, Gourlay CW, Dawes IW. Cellular redox homeostasis, reactive oxygen species and replicative ageing inSaccharomyces cerevisiae.FEMS Yeast Res, 2014, 14(1): 60–72.
[23] Ueda Y, Yonemitsu M, Tsubuku T, et al. Flavor characteristics of glutathione in raw and cooked foodstuffs. Biosci Biotechnol Biochem, 1997,61(12): 1977–1980.
[24] Wang B, Liang GB, Zhou QF, et al. Combined amino acids modulation with H2O2stress for glutathione overproduction inCandida utilis. Afr J Biotechnol, 2010, 9(33): 5399–5406.
[25] Wang DZ, Wang C, Wu H, et al. Glutathione production by recombinantEscherichia coliexpressing bifunctional glutathione synthetase. J Ind Microbiol Biotechnol, 2016, 43(1): 45–53.
[26] Zhang J, Greasham R. Chemically defined media for commercial fermentations. Appl Microbial Biotechnol,2004, 51: 407–421.
猜你喜欢
环球时报(2022-09-20)2022-09-20 15:18:57
酿酒科技(2021年8期)2021-12-06 15:28:22
军事文摘·科学少年(2021年1期)2021-02-04 08:03:45
今日农业(2020年24期)2020-12-15 16:16:00
中国调味品(2017年2期)2017-03-20 16:18:25
创新作文(小学版)(2016年16期)2016-11-11 05:47:54
现代检验医学杂志(2016年5期)2016-08-20 03:17:04
兽医导刊(2016年12期)2016-05-17 03:51:50
故事作文·低年级(2016年7期)2016-05-14 09:39:46
中国科技信息(2015年2期)2015-11-16 08:18:32
