
儿童Dent病6例临床特征及基因分析
2017-04-12 11:50:17孙建新,郝胜,孙利文等
临床儿科杂志 2017年1期
儿童Dent病6例临床特征及基因分析
孙建新1,2郝 胜1*孙利文1匡新宇1吴 滢1朱光华1何威逊1黄文彦1
1.上海市儿童医院 上海交通大学附属儿童医院肾脏风湿科(上海 200062);
2. 宁波市妇女儿童医院肾脏科(浙江宁波 315012)
目的探讨Dent病的早期诊断、治疗及预后。方法回顾性分析2013年1月至2015年12月确诊的6例Dent病患儿的临床资料。结果6例患儿均为男性,年龄3~10岁,均无氨基酸尿、尿糖、低钾,均有肾钙质沉着,2例有蛋白尿家族史,1例伴有镜下血尿,1例肾脏病理为局灶节段性肾小球硬化(FSGS),1例伴有佝偻病及肾功能不全;2例行基因检测示基因突变位点分别为c.731C>T(p.Ser244Leu)和c.848C>T(p.Pro283Leu);随访过程中6例患儿肾功能稳定。结论Dent病的典型症状为小分子蛋白尿、高钙尿,肾脏钙化等,基因诊断有助于早期确诊,早期诊断及治疗可改善预后。
小分子量蛋白尿; 高钙尿症; Dent病; CLCN5基因; 儿童
Dent病(Dent disease,DD)是一种罕见的X连锁遗传性肾小管功能障碍,其临床特点为小分子蛋白尿(low-molecular-weight proteinuria,LMWP)、高钙尿、肾钙质沉着或肾结石以及进行性肾功能减退,1964年由Dent等首先报道。Dent病患者典型的表现为近端肾小管功能障碍,包括LMWP、高尿磷、氨基酸尿、糖尿等,大约有30%~80%的男性患者在30~50岁之间发展成慢性肾功能不全[1]。随着国内对Dent病以及相关肾小管疾病认识的逐步加深以及临床诊断和基因诊断水平的不断提高,该病也逐渐引起临床医师的重视,现回顾性分析6例Dent病患者的临床资料。
1 临床资料
2013年1月至2015年12月在上海市儿童医院住院确诊为Dent病的6例患儿。Dent病诊断符合以下标准[2]:①小分子量蛋白尿(尿蛋白电泳证实的小分子蛋白尿);②高钙尿症,即24 h尿钙>0.1 mmol/kg或尿钙/肌酐比>0.25;③至少有下列情况之一,肾钙质沉着、肾结石、血尿、低磷血症或肾功能不全。
6例Dent病患儿均为男性,年龄4~13岁,起病年龄3~10岁,从起病到确诊均在2个月之内(表1),其中例3在外院诊断5年后转至我院随访。6例患儿均无氨基酸尿、尿糖、低钾、高脂血症,均有肾钙质沉着;例3因伴有大量蛋白尿行肾活检术病理提示局灶节段性肾小球硬化(focal segmental glomerular sclerosis,FSGS);2例有蛋白尿家族史,1例伴有镜下血尿。
?
6例患儿中5例为体检或因其他原因尿检时发现蛋白尿,均无明显主诉症状,另外1例患儿因“O”型腿在骨科行矫正手术前检查时发现,也无除佝偻病以外的其他临床表现。
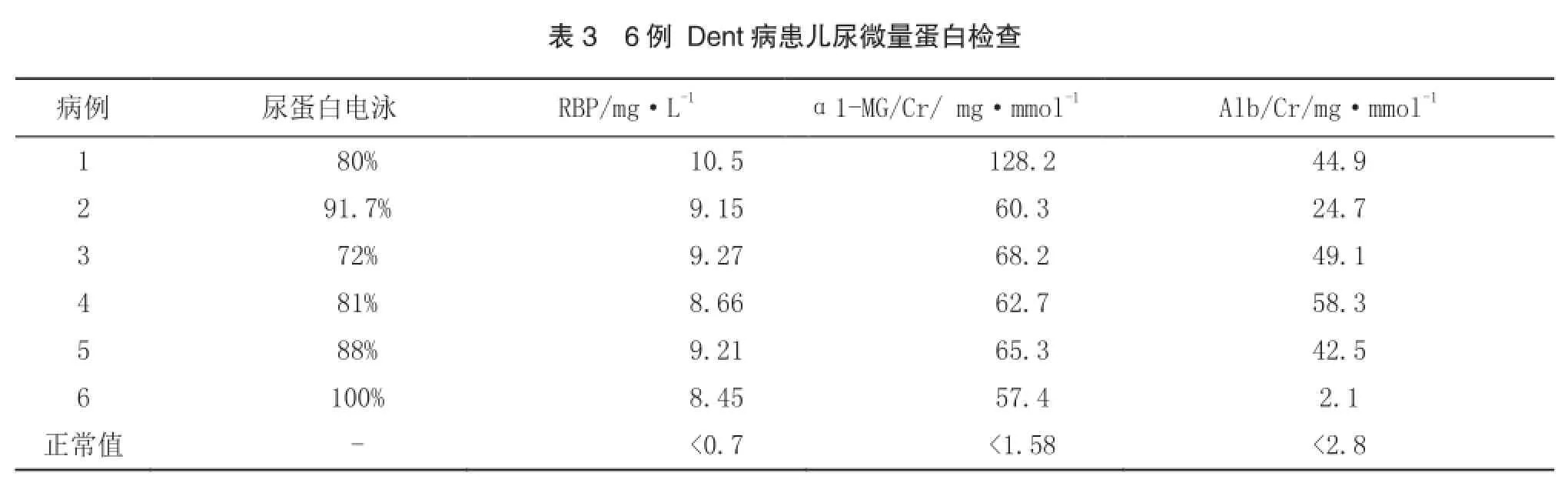
6例患儿均有典型的高钙尿和小分子蛋白尿,包括蛋白电泳显示小分子蛋白尿为主,尿系列微量蛋白中α1-MG、视黄醛结合蛋白(RBP)明显升高(超过正常值5倍),5例患儿尿白蛋白也升高,6例患儿中仅1例血清肌酐升高伴血磷轻度降低,其余5例肾功能正常。见表2,3。
3例行基因检测均发现为CLCN5基因突变。例1的基因检测方法为1代测序,发现其基因编码序列第731位碱基C变为T(c.731C>T),氨基酸改变为p.Ser244Leu(图1);例2的基因检测方法为2代基因测序技术,发现其基因编码序列第848位碱基C变为T(c.848C>T),氨基酸改变为p.Pro283Leu,为半合子突变,父无突变(图2);两例患儿母亲为杂合携带者,突变均为错义突变。另1例为外院基因检测。
明确诊断的患儿均予氢氯噻嗪对症治疗,24 h蛋白尿>1 g的给予血管紧张素Ⅱ受体拮抗剂(angiotensinⅡ receptor antagonist,ARB)治疗,例1由于佝偻病及肾功能不全给予骨化三醇治疗,例2由于肾髓质钙盐沉积明显给予枸橼酸碱化尿液治疗。6例患儿中,例3在外院确诊后在我院随访3年,故最长随访时间达8年。随访过程中,例1肾功能较前明显改善,另外5例患儿均肾功能正常。
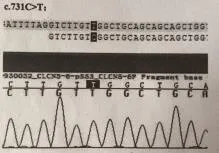
图1 例1患儿CLCN5基因测序图
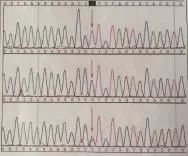
图2 例2患儿及父母CLCN5基因测序图
2 讨论
蛋白尿是儿童肾脏疾病最常见的临床表现之一,通过对尿蛋白成分的检测,可以初步明确其来源于肾小球或肾小管,从而进一步确定肾脏病变的性质。近年来,由于对肾小管疾病的重视程度提高,以及肾脏病理和基因技术的发展,对肾小管疾病的了解更加深入。以LMWP和高钙尿症为主要表现的Dent病是近年来备受重视的肾小管疾病之一[1,3,4]。Dent病主要表现为近端小管功能障碍, 典型Dent病的诊断符合前述标准,并可发生并发症如佝偻病和软骨病等[2]。本组6例患儿符合低分子蛋白尿、高钙尿,伴有双肾结晶或钙化,其中1例因佝偻病就诊而确诊,且已发展至肾功能不全。由于LMWP是Dent病的首要诊断条件,检测分析尿蛋白的成分对其有重要意义。传统的方法主要是尿蛋白电泳,通过其分子量的大小确定其蛋白尿的成分,虽然不能检测具体的蛋白质种类,但能较直观的判断是否存在LMWP。本组6例患儿的尿蛋白电泳均能明确显示尿液中LMWP占主要比例,因此对Dnet病的诊断有重要意义。尽管目前各种关于Dent病的诊断标准均以其临床特点为要素,内容也是大同小异,国外一些文献中将LMWP定义为单纯的RBP或α1-MG或β2-MG升高,本组病例也符合RBP和α1-MG升高5倍的标准,而临床工作中大部分肾小球疾病也存在一定量的LMWP,甚至会超过正常值的5倍以上,但仍以中大分子蛋白为主,而本组病例中6例患儿有5例也存在一定量的小球蛋白,如果单纯以尿系列微量蛋白鉴别极易出现误诊,因此笔者认为诊断标准中应将传统的尿蛋白电泳列为确定LMWP的主要方法之一。除此之外,诊断标准中还提到除了LMWP和高钙尿以外,至少还需要具备肾钙质沉着、肾结石、血尿、低磷血症或肾功能不全等表现,随着对疾病的认识加深以及基因诊断的发展,本着早发现早治疗的原则,Dent病的诊断是否必须要有上述的一些表现有待商榷。随着人类基因诊断技术的发展,人们逐渐认识到Dent病主要是CLCN5和OCRL1基因突变所致,基因诊断也越来越多地应用于临床,已有文献将基因诊断列为Dent病的必要诊断标准之一[5]。
?
?
Dent病通常发生在男性,可在儿童早期出现,女性携带者症状轻微,可仅有镜下血尿,发病率还不清楚,目前约250个家庭的病例已被报道[6]。本组6例患儿均为男性。目前已知约有60%的Dent病是由CLCN5基因突变所致,被命名为1型Dent病(孟德尔遗传数据库OMIM300009)[7,8],CLCN5基因位于染色体Xp11.22-p11.23,编码由746个氨基酸组成的Cl-通道蛋白-5(chloride channel-5,CLC-5),包含17个外显子和来自至少4个不同起始位点的转录启动子,这些外显子和转录启动子在翻译转录过程中发生的多种变异可导致CLCN5基因突变,现已有200多种基因突变被报道[9]。CLC-5属于电压门控性氯离子通道蛋白家族成员,主要在近端肾小管、皮质集合小管和髓袢升支粗段表达,在肝脏、心脏、结肠等组织中也有表达。CLC-5在肾小管上皮细胞的内涵体中,通过促进Cl-内流中和H+-ATPase泵出的H+对控制细胞膜的兴奋性以及内涵体中的酸化过程起着重要作用。也有学者通过研究证实, Cl-浓度在胞吞作用中比酸化作用更加重要[10],而CLC-5被认为是一种2Cl-/H+交换体,而不仅仅是Cl-通道[6]。CLCN5基因突变可导致CLC-5通道蛋白结构异常和功能障碍,Cl-内流受限,内涵体及溶酶体酸化过程受阻,从而引起胞吞作用障碍,出现维生素D结合蛋白、25羟维生素D3和甲状旁腺激素等小分子蛋白尿[11,12]。近年来研究显示编码具有磷脂酶活性OCRL1蛋白(磷脂酰肌醇4,5-二磷酸5-磷酸酶)的OCRL1基因突变也可导致类似Dent病1的临床表现,在OMIM数据库中被命名为2型Dent病 (OMIM 300555),OCRL1基因最早被发现是Lowe综合征(眼-脑-肾综合征)的致病基因[13],文献报道约15%的典型Dent病临床表现的患者存在OCRL1基因突变[8,14,15]。由于OCRL1基因普遍存在于人体组织中,包括眼睛、肾脏和大脑,故由OCRL1基因突变引起的2型Dent病可出现肾外症状[16],但不同于同样由OCRL1基因突变引起的Lowe综合征,2型Dent病患者无典型的眼睛和中枢神经系统异常,可能与基因突变的位点不同有关。目前,仍有25%~35%的Dent病患者既无CLCN5基因突变亦无OCRL1基因突变,故可能存在其他基因的突变,也有两个基因共同致病的报道[17]。本组病例中3例行基因检测均为CLCN5基因突变所致,其中在上海市儿童医院检测的2例突变位点分别为c.731C>T,p.Ser244Leu和c.848C>T,p.Pro283Leu,为已明确的基因突变,但更多的病例及突变位点仍有可能不断出现,随着基因检测的逐渐普及,包括Dent病、Alport综合征、Fanconi综合征、Batter综合征在内的先天/遗传性疾病的诊断越来越多的依赖于基因诊断,但家系的验证、基因型和表型之间仍存在着较多目前无法解释的问题,因此临床诊断仍应放在疾病诊治的首要位置。除此之外,对于这些罕见疾病有必要通过全国的注册系统将其临床资料、突变位点、家系的情况进行详细整理,以提高对这类疾病的诊治和管理水平。
虽然Dent病是肾小管病变,但也会伴有肾小球性疾病,有报道Dent病伴有肾病水平蛋白尿可能存在肾小球损害,肾活检发现有局灶性肾小球硬化[15]。本组1例患儿因大量蛋白尿行肾活检提示为FSGS,提示部分Dent病患者可能还同时存在肾小球病变,其发病机制并不清楚。此外,本组另外3例患者因各种原因未行基因检测,2例有肾脏疾病家族史,其中1例伴有血尿,其胞姐和父亲有蛋白尿(胞姐肾脏病理为轻微病变),另1例患儿外婆有慢性肾炎病史,其间的联系有待进一步随访观察。
由于Dent病是一种先天遗传性疾病,目前无法治愈,以对症治疗为主,主要是降低尿钙、预防肾结石的形成和肾脏钙化,延缓肾间质纤维化和肾功能恶化。噻嗪类利尿药物是降低尿钙的首选,但长时间大剂量应用有一定风险,维生素D可预防和治疗佝偻病,枸橼酸盐碱化尿液对于预防肾结石和延缓肾功能恶化有一定作用。本组6例患儿随访中仍有蛋白尿,程度有所减轻,而肾功能均较稳定,但由于前述提到的部分男性患者可在25~50岁随时进展至肾功能不全,因此是否会有进展性的肾功能衰竭仍需长期的随访。Dent病的早期诊断、早期治疗对于延缓肾功能恶化至关重要,本组1例伴有FSGS的患儿已经明确诊断及随访8年,肾功能稳定。由于Dent病的临床表现多样,而且部分患儿症状轻微,生化指标改变不明显,以至于误诊、漏诊,故对儿童常规的尿筛查也可起到早期发现肾脏疾病的积极作用。
总之,Dent病是遗传性肾小管疾病,大部分是CLCN5、OCRL1基因突变引起,典型症状为低分子蛋白尿、高钙尿,肾脏钙化等,基因诊断对疾病的确诊有意义,早期诊断及降低尿钙治疗可减轻肾脏钙化和肾小管间质纤维化程度,从而延缓其终末期肾病的进展。
[1] Anglani F, D’Angelo A, Bertizzolo LM, et al. Nephrolithiasis, kidney failure and bone disorders in Dent disease patients with and withoutCLCN5mutations [J]. Springerplus, 2015, 4:492.
[2] Hoopes RR Jr, Raja KM, Koich A, et al. Evidence for genetic heterogeneity in Dent's disease [J]. Kidney Int, 2004, 65(5):1615-1620.
[3] 朱碧溱,李鹏,黄建萍.以小分子蛋白尿为主要表现的Dent病六例临床及基因分析[J].中华儿科杂志, 2010, 48(5): 329-333.
[4] 张宏博, 黄建萍. 药物治疗Dent 病15例的临床疗效[J].中华实用儿科临床杂志, 2016(3): 226-230.
[5] Tang X, Brown MR, Cogal AG, et al. Functional and transport analyses ofCLCN5genetic changes identified in Dent disease patients [J]. Physiological Reports, 2016, 4(8):e12776.
[6] Wu F, Reed AA,Williams SE,et a1. Mutalional analysis of CLC-5, cof i lin and CLC-4 in patients with Dent’s disease [J]. Nephron Physiol, 2009, l12(4): 53-62.
[7] Claverie-Martin F, Ramos-Trujillo E, Garcia-Nieto V. Dentˊs disease: clinical features and molecular basis [J]. Pediatr Nephrol, 2011, 26(5) : 693-704.
[8] Ludwig M, Levtchenko E, Bökenkamp A. Clinical utility gene card for: Dent disease (Dent-1 and Dent-2) [J]. Eur J Hum Genet, 2014, 22(11). doi: 10.
[9] 张宏博, 黄建萍. 儿童Dent病研究进展[J]. 中国实用儿科杂志, 2015(1): 77-80.
[10] Novarino G, Weinert S, Rickheit G, et al. Endosomal chloride-proton exchange rather than chloride conductance is crucial for renal endocytosis [J]. Science, 2010, 328(5984):1398-1401.
[11] Lippiat JD, Smith AJ. The CLC-5 2Cl( -) / H( + ) exchange transporter in endosomal function and Dent’s disease [J]. Front Physiol, 2012, 3: 449.
[12] Tosetto E, Casarin A, Salviati L, et al. Complexity of the 5ˊUTR region of theCLCN5gene: eleven 5ˊUTR ends are differentially expressed in the human kidney [J]. BMC Med Genomics, 2014, 7(1): 41.
[13] Bökenkamp A, Levtchenko E, Recker F, et al. Clinical utility gene card for: Lowe syndrome [J]. Eur J Hum Genet, 2015, 23(6). doi: 10.1038/ejhg.
[14] Grand T, L'Hoste S, Mordasini D, et al. Heterogeneity in the processing ofCLCN5mutants related to Dent disease[J]. Hum Mutat, 2011, 32(4) : 476-483.
[15] Cramer MT, Charlton JR, Fogo AB, et al. Expanding the phenotype of proteinuria in Dent disease. A case series [J]. Pediatr Nephrol, 2014, 29(10) : 2051-2054.
[16] Shrimpton AE, Hoopes RR Jr, Knohl SJ, et a1. OCRLl mumtions in Dent 2 patients suggest a mechanism for phenotypic variability [J]. Nephron Physiol, 2009, 112(2): 27-36.
[17] Addis M, Meloni C, Tosetto E et al. An atypical Dent's disease phenotype caused by co-inheritance of mutations atCLCN5andOCRLgenes [J]. Eur J Hum Genet,2013 21(6):687-690.
Clinical features and genetic analysis of Dent disease in 6 children
SUN Jianxin1,2, HAO Sheng1, SUN Liwen1, KUANG Xinyu1, WU Ying1, ZHU Guanghua1, HE Weixun1, HUANG Wenyan1(1.Department of Nephrology and Rheumatology, Children’s Hospital of Shanghai, Children’s Hospital of Shanghai Jiaotong University, Shanghai 200040, China; 2. Department of Nephrology, Ningbo Women and Children Hospital, Ningbo 315012, Zhengjiang, China)
ObjectiveTo explore the early diagnosis, treatment and prognosis of Dent disease.MethodThe clinical data of 6 children with Dent disease were retrospectively analyzed from January 2013 to December 2015.Resultsin 6 male chilren aged 3-10 years old, all of the children didn't have amino acid urine, urine glucose, or low potassium, while suffered with nephrocalcinosis. Two children had family history of proteinuria, one case was combined with microscopic hematuria, one showed glomerular focal segmental sclerosis (FSGS) in renal pathology, and one case accompanied with rickets and renal insuff i ciency. Two children were performed the gene detection and it showed the gene mutation site at c.731C>T (p.Ser244Leu) and c.848C>T(p.Pro283Leu) respectively. During the follow-up, the renal function was stable among all of the children.ConclusionsThe classical symptoms of Dent disease were low molecular weight proteinuria, hypercalcinuria, renal calcif i cation, and so on. Gene detection is helpful in early diagnosis. Early diagnosis and treatment can improve the prognosis.
low weight proteinuria; hypercalcinuria; Dent disease;CLCN5gene; child
10.3969/j.issn.1000-3606.2017.01.015
2016-07-17)
(本文编辑:梁 华)
黄文彦 电子信箱:hwy65@hotmail.com
*共同第一作者
猜你喜欢
英语世界(2023年6期)2023-06-30 06:29:10
昆明医科大学学报(2021年12期)2021-12-30 06:59:44
中国民间疗法(2021年19期)2021-11-20 06:22:34
中国民间疗法(2021年18期)2021-11-02 08:20:16
中国生殖健康(2020年2期)2021-01-18 02:51:26
小学生导刊(2018年13期)2018-06-29 03:49:00
中国继续医学教育(2015年2期)2016-01-06 01:36:29
中外医疗(2015年11期)2016-01-04 03:58:45
医学研究杂志(2015年8期)2015-06-22 14:00:57
医学研究杂志(2015年12期)2015-06-10 06:57:46
