
星点设计-效应面法优化萆薢分清饮超声-微波协同提取工艺研究*
2017-04-10 06:47:08叶颖俊徐子金陈慧刘建明邱模昌
世界科学技术-中医药现代化 2017年2期
叶颖俊,徐子金,陈慧,刘建明,邱模昌
(江西医学高等专科学校药学系上饶334000)
星点设计-效应面法优化萆薢分清饮超声-微波协同提取工艺研究*
叶颖俊,徐子金**,陈慧,刘建明,邱模昌
(江西医学高等专科学校药学系上饶334000)
目的:采用星点设计-效应面法,优选萆薢分清饮超声-微波协同提取工艺,并与传统水煎煮工艺进行对比。方法:分别考察微波频率、加水倍量、提取时间等主要因素,将薯蓣皂苷元、甘草酸提取率及浸膏收率作为评价指标,以综合评分为响应值作响应面和等高线,进行预测分析,获得最优提取条件。结果:确定萆薢分清饮的最优提取工艺:微波频率为434W,加水倍量为18.4倍,提取时间为9.3min,超声频率固定为50W。采用该提取工艺,薯蓣皂苷元的提取收率为23.17%(mg·g-1),甘草酸的提取收率为0.64%(g·g-1),浸膏的提取收率为34.12%(g·g-1),各项指标均高于传统水煎煮工艺。结论:星点设计-效应面法适用于萆薢分清饮的提取工艺优化,建立的数学模型预测性好,优化所得的超声-微波协同提取工艺操作简便,提取效率高,适用于工业化生产,同时也为萆薢分清饮的现代制剂研发提供科学理论基础。
萆薢分清超声-微波协同星点设计提取率
“萆薢分清”一词最早载于《杨氏家藏方》,该方由川萆薢、益智仁、石菖蒲、乌药组成,有温肾利湿,分清化浊之功用。后世医家在《丹溪心法》中记载“每服五钱,水煎,入盐一捻,食前服。一方加茯苓、甘草”。方中川萆薢为君药,善于利湿,分清化浊,是治白浊之要药。益智仁温肾阳,缩小便,为臣药。乌药温肾祛寒,暖膀胱以助气化;石菖蒲芳香化浊,分利小便,共为佐药。2015年版《中国药典》[1]收载的成方制剂萆薢分清丸在古方的基础上,以粉萆薢替代川萆薢,加甘草调和诸药,主治肾不化气、清浊不分所致的白浊、小便频数。萆薢分清丸将生药材直接粉碎,用水泛丸,干燥后以滑石粉包衣、打光即得。由于药物未经提取,患者单次用量较大,临床应用顺应性较差。随着中药现代化研究的不断推进,以中药物质基础为理论的“量-效”关系成为研究的热点之一[2]。采用适宜的工艺将中药复方进行提取,并进一步制成药物制剂新剂型,既有利于古方的物质基础研究,同时也有助于临床应用的减毒增效。
超声波和微波作为现代化提取技术,与传统溶剂提取法相比,具有提取间短、提取效率高、能耗低等优点,在中药提取中广泛应用[3-8]。星点设计是国内外常用的实验设计方法之一,广泛应用于处方筛选、工艺考察等方面,具有精密度高、试验次数少、预测性好以及重现性好等多方面的优点[9]。效应面法与星点设计相结合,通过建立数学模型,使得试验结果直观立体,工艺参数筛选更加精密准确。本试验采用超声-微波协同提取技术,通过星点设计-效应面法优化萆薢分清饮的提取工艺,描述提取工艺参数与有效成分收率之间的数量关系,为萆薢分清饮的现代研究提供一定的依据,同时也为该方法在中药复方提取中的应用提供一定的理论基础。
1 仪器与试药
1.1 仪器
CW-2000超声-微波协同萃取仪(上海新拓分析仪器科技有限公司);1260型高效液相色谱仪,自带Chemstation A.01.04色谱工作站,G1314F紫外检测器(美国安捷伦科技公司);BL-500万能高速粉碎机(德州市昊诚实验仪器有限公司);Al204型电子天平(上海梅特勒-托利多仪器有限公司);GKC-11-CR2电热恒温水浴箱(上海科技试验仪器厂);KQ3200DB型数控超声波清洗器(昆山市超声仪器有限公司)。
1.2 试药
薯蓣皂苷元对照品(纯度为100%,中国食品药品检定研究院,批号:111539-200001);甘草酸铵对照品(纯度为93.0%,中国食品药品检定研究院,批号:110731-201619);粉萆薢、益智仁、石菖蒲、乌药、甘草均购自上饶市中医院药房,经江西医学高等专科学校生药教研室汪亮副教授鉴定,粉萆薢为薯蓣科植物粉背薯蓣的干燥根茎;益智仁为姜科植物益智的干燥成熟果实;石菖蒲为天南星科植物石菖蒲的干燥根茎;乌药为樟科植物乌药的干燥块根;甘草为豆科植物甘草的干燥根茎;所用中药饮片均被2015版《中国药典》所记录。乙腈、甲醇(色谱纯,美国Sigma公司),自制纯化水,其他所有试剂均为分析纯。
2 方法与结果
2.1 薯蓣皂苷元含量测定方法的建立
2.1.1 色谱条件[10]
色谱柱为Diamonsil C18(250mm×4.6mm,5μm);流动相为甲醇:乙腈(30:70);流速1 mL·min-1,柱温30℃,检测波长206 nm,进样量10μL。理论板数按薯蓣皂苷元峰计算应不低于4 000。在该色谱条件下,薯蓣皂苷元色谱峰与其他色谱峰分离良好(图1)。
2.1.2 对照品溶液的制备
精密称取干燥至恒重的薯蓣皂苷元对照品适量,加适量甲醇溶解后,稀释至浓度为1mg·mL-1对照品储备液。精密量取对照品储备液适量,加甲醇稀释至浓度为0.2mg·mL-1,即得对照品溶液。
2.1.3 供试品溶液的制备
按处方量称取各味药材,粉碎后过40目筛网,照星点设计试验表进行提取试验,将提取液过滤,滤液合并后浓缩至250mL,得供试品储备液(每1mL相当于0.66 g生药材)。精密量取10mL供试品储备液,加2mol·L-1的盐酸溶液50mL,水浴回流2 h,取出,放冷,转移至分液漏斗中,加氯仿进行萃取,每次20mL,共萃取3次,合并萃取液,回收溶剂至干,残渣加适量甲醇溶解至10mL容量瓶中,定容,用0.45μm的微孔滤膜过滤,得续滤液即供试品溶液。
2.1.4 阴性对照溶液的制备
按照处方制备不含萆薢的样品,按“2.1.3”项下供试品溶液的制备的方法制备阴性对照溶液,在上述色谱条件下测定,与对照品溶液和供试品溶液比对,结果表明在薯蓣皂苷元相应保留时间处无吸收峰,表明其它成分不干扰薯蓣皂苷元的含量测定(图1)。
2.1.5 线性关系考察
分别精密吸取对照品储备液适量,加甲醇稀释,制得0.102、0.204、0.408、0.612、0.816 mg·mL-1标准溶液。取10μL进行HPLC分析,记录色谱峰面积,以溶液浓度为横坐标,以峰面积为纵坐标绘制标准曲线。制得薯蓣皂苷元标准曲线,回归方程为:Y=2.365× 106X+3.977×105,r=0.999 6,实验结果表明薯蓣皂苷元在0.102-0.816mg·mL-1呈良好的线性关系。
2.1.6 精密度试验
精密吸取对照品溶液10μL连续重复进样6次,记录薯蓣皂苷元色谱峰面积,峰面积RSD值为1.08%,表明仪器精密度良好。
2.1.7 稳定性试验
精密吸取对照品溶液和供试品溶液10μL分别于制备后0、1、2、4、6、8、12 h进行测定,记录峰面积值,对照品溶液的RSD为0.94%,供试品溶液的RSD为1.26%,表明对照品溶液和供试品溶液在12 h内稳定。
2.1.8 重复性试验
取同一批供试品储备液,按“2.1.3”项下供试品溶液制备方法制备供试品溶液6份,按上述色谱条件进行含量测定,记录得峰面积值,薯蓣皂苷元重复性试验的RSD%为1.42%,表明该方法重现性良好。
2.1.9 加样回收率试验
精密吸取已知含量的供试品储备液各9份,精密加入低、中、高三个浓度的薯蓣皂苷元对照品,按照“2.1.3项下”供试品溶液制备方法制备供试品溶液,按上述色谱条件进行测定,计算加样回收率,平均回收率为98.09%,RSD%为0.56%,表明方法准确性良好。
2.1.10含量测定
取试验所得供试品溶液,照上述色谱条件检测,计算薯蓣皂苷元的提取收率,收率计算公式:
薯蓣皂苷元收率=储备液中薯蓣皂苷元总量(mg)/提取药材量(g)×100%
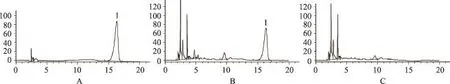
图1 萆薢分清饮各溶液的高效液相色谱图
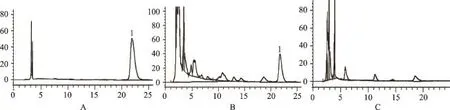
图2 各溶液的高效液相色谱图
2.2 甘草酸含量测定方法的建立
2.2.1 色谱条件
色谱柱为Diamonsil C18(250mm×4.6mm,5μm);流动相为甲醇:0.2mol·L-1醋酸铵溶液:冰醋酸(56:43:1);流速1mL·min-1,柱温30℃,检测波长250 nm,进样量10μL。理论板数按甘草酸峰计算应不低于5 000。在该色谱条件下,甘草酸色谱峰与其他色谱峰分离良好(图2)。
2.2.2 对照品溶液的制备
精密称取干燥至恒重的甘草酸铵对照品适量,加适量流动相溶解后,稀释至浓度为1mg·mL-1,作为对照品储备液。精密量取对照品储备液适量,加流动相稀释至浓度为0.2mg·mL-1,即得对照品溶液。
2.2.3 供试品溶液的制备
按处方量称取各味药材,照星点设计试验表进行提取试验,将提取液过滤,滤液合并后浓缩至250mL,得供试品储备液(每1mL相当于0.66 g生药材)。精密量取10mL供试品储备液,精密加入流动相50mL,称定重量,加热回流1 h,取出,放冷,再称定重量,用流动相补足减失的重量,摇匀,用0.45μm的微孔滤膜过滤取续滤液即得供试品溶液。
2.2.4 阴性对照溶液的制备
按照处方制备不含甘草的样品,按“2.2.3”项下供试品溶液的制备的方法制备阴性对照溶液,在上述色谱条件下测定,与对照品溶液和供试品溶液比对,结果表明在甘草酸相应保留时间处无吸收峰,表明其它成分不干扰甘草酸的含量测定(图2)。
2.2.5 线性关系考察
分别精密吸取对照品储备液适量,加流动相稀释,制备0.052、0.104、0.208、0.416、0.624、0.832、1.04mg·mL-1标准溶液。取标准溶液10μL注入液相色谱仪,记录色谱峰面积,以溶液浓度为横坐标,以峰面积为纵坐标绘制标准曲线。制得甘草酸标准曲线,回归方程为:Y=5.24×106X-2.41×103,r=0.999 5,实验结果表明:甘草酸在0.052-1.040mg·mL-1呈良好的线性关系。
2.2.6 精密度试验
精密吸取对照品溶液10μL连续重复进样6次,记录甘草酸色谱峰面积,峰面积RSD值为1.13%,表明仪器精密度良好。
2.2.7 稳定性试验
精密吸取对照品溶液和供试品溶液10μL分别于制备后0、1、2、4、6、8、12 h进行测定,记录峰面积值,对照品溶液的RSD为1.20%,供试品溶液的RSD为1.37%,,表明对照品溶液和供试品溶液在12 h内稳定。
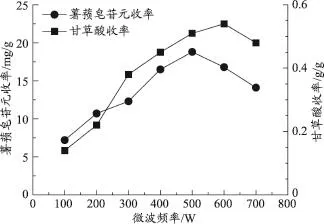
图3 微波频率对薯蓣皂苷元和甘草酸收率的影响
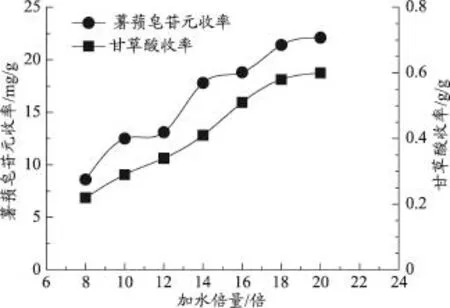
图4 加水倍量对薯蓣皂苷元和甘草酸收率的影响
2.2.8 重复性试验
取同一批供试品储备液,按“2.2.3”项下”供试品溶液制备方法制备供试品溶液6份,按上述色谱条件进行含量测定,记录得峰面积值,甘草酸重现性试验的RSD%为0.97%,表明该方法重复性好。
2.2.9 加样回收试验
精密吸取已知含量的供试品储备液各9份,精密加入低、中、高三个比例的甘草酸铵对照品,按照“2.2.3”项下供试品溶液制备方法制备供试品溶液,按上述色谱条件进行测定,计算加样回收率,平均回收率为98.18%,RSD%为1.01%,表明改方法准确性好。
2.2.10含量测定
取各试验所得供试品溶液,照上述色谱条件检测,计算甘草酸的提取收率。收率计算公式如下:
甘草酸收率=储备液中甘草酸总量(g)/提取药材量(g)×100%
2.3 干浸膏量的测定
精密量取浓缩液10mL,分别置于重量恒定的蒸发皿,水浴蒸干,然后转移至105℃下烘干至恒重,计算干浸膏得率。
2.4 星点设计法优化超声-微波协同提取工艺
2.4.1 选择提取因素及相关水平
(1)微波功率范围的选择
准确称取处方量的药材,粉碎后过40目筛,在加水倍量为16倍、提取时间为10min、超声功率固定为50W的条件下,研究不同微波频率(100、200、300、400、500、600、700W)对薯蓣皂苷元和甘草酸提取收率的影响。
由图3可知,薯蓣皂苷元和甘草酸的提取收率随着微波频率的增大,呈现先增大后减小的趋势。薯蓣皂苷元的提取收率在微波频率为500W时达到最大值。甘草酸的提取收率在微波频率为600W时达到最大值,但在此微波频率下,薯蓣皂苷元的提取收率下降明显,可能是微波产生的热效应对薯蓣皂苷元有一定的破坏作用,因此确定微波频率的考察范围为100-500W。
(2)加水倍量范围的选择
准确称取处方量的药材,粉碎后过40目筛,在微波频率为500W,提取时间为10min,超声功率固定为50W的条件下,研究不同加水倍量(8、10、12、14、16、18、20倍)对薯蓣皂苷元和甘草酸提取收率的影响。
由图4可知,薯蓣皂苷元和甘草酸的提取收率随着加水倍量的增大而逐渐增大。当加水倍量达到20倍时,随着加水倍量的增加,薯蓣皂苷元和甘草酸的提取收率变化平缓,因此,确定加水倍量的考察范围为8-20倍。
(3)提取时间范围的选择
准确称取处方量的药材,粉碎后过40目筛网,在微波频率为500W,加水倍量为16倍,超声功率固定为50W的条件下,研究不同提取时间(2、4、6、8、10、12、14、16、18、20min)对薯蓣皂苷元和甘草酸提取收率的影响。
图5表明,薯蓣皂苷元和甘草酸的提取收率随着提取时间的增加,呈现先增大后减小的趋势。当提取时间达到14min时,随着提取时间的增加,薯蓣皂苷元和甘草酸的提取收率变化平缓,且略有下降,因此,确定提取时间的考察范围为2-14min。
2.4.2 星点试验设计及结果
根据单因素试验考察结果,以微波频率(X1)、加水倍量(X2)和提取时间(X3)作为考察因素,因素的取值范围如下:100≤X1≤500,8≤X2≤20,2≤X3≤14。以薯蓣皂苷元、甘草酸、及浸膏收率作为考察指标,通过星点设计试验进一步优选提取工艺。数据处理采用“归一化法”,采用“Hassan法”[11]将每个指均转化为0-1之间的“归一值”,并计算几何平均数,得综合评分。计算公式:
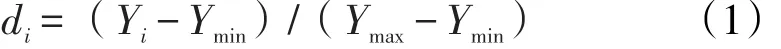
Yi为实测值,Ymin和Ymax分别指的该指标在所有试验中的最小值和最大值。将所计算得到的di值带入公式2,即得综合评分Y值。
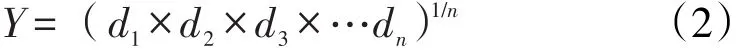
根据星点设计的原理,各因素设置5水平,因素及水平见表1,实验安排及效应值见表2。
2.4.3 星点设计模型拟合
以SPSS 17.0为统计软件,以综合评分Y对各因素进行多元线性拟合,拟合方程:
Y=-1.130 217+1.641×10-3X1+7.113 3×10-2X2
+2.350 2×10-2X3(r=0.970 6,P<0.05)。
以综合评分对各因素进行二次多项式拟合,拟合方程:
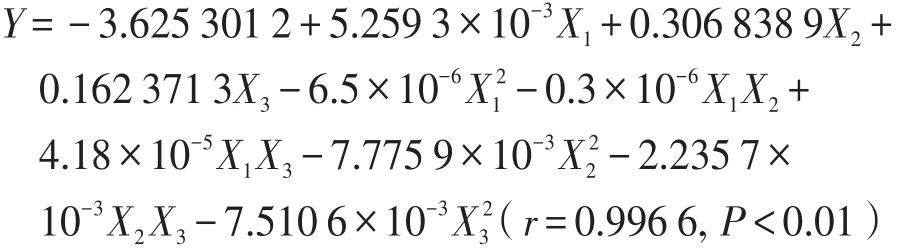
根据拟合方程的拟合优度(r)和置信度(P)可知,二次多项式方程的拟合结果优于多元线性方程拟合结果,其拟合结果可信度高,根据P值可知,微波频率、加水倍量、提取时间均具有显著性差异,二次多项式各系数值见表3。
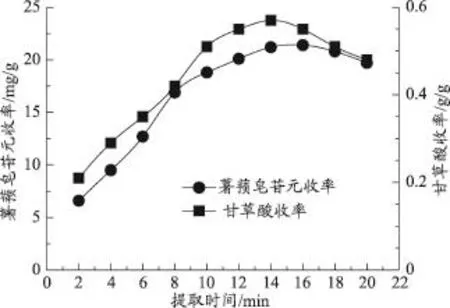
图5 提取时间对薯蓣皂苷元和甘草酸收率的影响
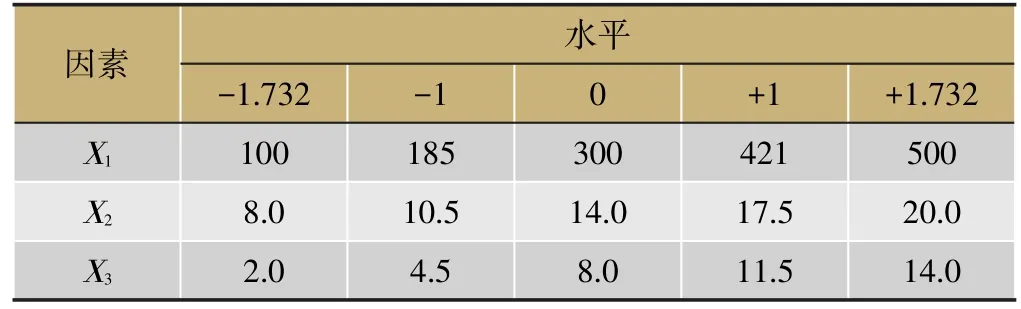
表1 因素各水平代码值及实际操作物理量

表2 试验设计安排及效应值表
2.4.4 拟合结果效应面优化及预测
固定三个自变量之一并取中间值,以拟合的目标函数为数学模型,采用Oringin 8.0统计软件绘制效应值对应自变量的效应面和等高线图。根据效应面图及二维等高线图可知,微波频率、加水倍量及提取时间3个因素中任意两个因素对综合评分的影响均较为显著,响应面图陡峭。为确定这3个因素的最佳取值,通过Design-Expert7.0软件分析,得出回归模型的最大值点,与之相对应微波频率为434W,加水倍量为18.4倍,提取时间为9.3min,超声频率固定为50W,结果见图6、图7、图8。

表3 二次多项式回归方程相关系数显著性检验

图6 二次多项式模型中微波频率、加水倍量对综合评分拟合效应面图及等高线图

图7 二次多项式模型中微波频率和提取时间对综合评分拟合效应面图及等高线图
2.4.5 验证及对比试验
为了验证所得到的提取模型的适用性,在微波频率、加水倍量、提取时间最优的水平上,重复试验3次。同时,与文献中的方法(每次加10倍量的水,煎煮3次,每次1 h)进行提取收率的对比[12]。验证试验中,薯蓣皂苷元的平均提取率为23.17%(mg·g-1),RSD为0.59%;甘草酸的平均提取率为0.64%(g·g-1),RSD为0.91%;浸膏平均收率为34.12%(g·g-1),RSD为0.62%。水煎煮提取试验中,薯蓣皂苷元的平均提取率为20.45%(mg·g-1),甘草酸的平均提取率为0.52%(g·g-1),浸膏平均收率为26.41%(g·g-1)。结果表明,采用超声-微波协同提取工艺,薯蓣皂苷元、甘草酸及浸膏收率均大于水煎煮提取。且从工艺简便程度、耗时耗能等多方面考虑,超声-微波协同提取工艺均具有一定的优势。
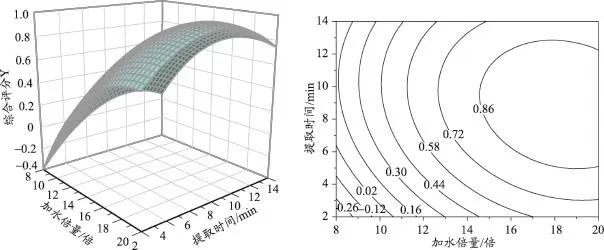
图8 二次多项式模型中加水倍量和提取时间对综合评分拟合效应面图及等高线图
3 讨论
微波能使药材产生强烈的内热效应,内部和外部同时加热,加速细胞的破裂,极大的提高有效成分的解吸附和溶解效率,快速进入提取溶剂中。超声提取技术的原理是利用超声的空化效应、热效应和机械效应,同时作用于细胞的破裂阶段、有效成分的溶解和扩散阶段[13]。
本试验联用微波和超声提取技术,在单因素试验的基础上,采用星点设计-效应面法,开发的萆薢分清饮协同提取工艺,从提取溶剂用量、提取时间、提取次数等方面考虑,降低了提取过程中的能耗,且工艺简便,提取率高,重现性好。
萆薢分清饮作为经典古方之一,临床上主要应用于慢性前列腺炎的治疗[14]。其中君药萆薢在泌尿系统疾病及肿瘤的治疗也有广泛应用[15]。采用现代科学技术,对该方剂进一步的研究与开发,有一定的理论和现实意义。本试验采用区别于传统水煎煮工艺的超声-微波协同提取法,对该方剂进行了提取工艺的研究,并进一步对其现代制剂工艺及质量控制标准进行研究。但是,本试验确定的方法仅为批量较小的实验室方法,由于微波从介质的表面进入并在其内部传播时,能量不断被吸收并转化为热能,其所携带的能量随着深入介质表面的距离而衰减。因此,本课题组将进一步对批量扩大后工艺参数变化规律进行研究。
1国家药典委员会.中华人民共和国药典(一部).北京:中国医药科技出版社,2015.
2肖平,李祥,陈建伟,等.中药药效物质基础研究思路与方法概述.时珍国医国药,2015,25(8):1935-1937.
3 Hossain M B,Brunton N P,Patras A,et al.Optimization of ultrasound assisted extraction of antioxidant compounds frommarjoram(Origanum majorana L)using response surface methodology.Ultrason Sonochem, 2012,19:582-590.
4 Wang X S,Wu Y F,Chen GY,etal.Optimisation of ultrasound assisted extraction of phenolic compounds from Sparganiirhizome with response surfacemethodology.Ultrason Sonochem.,2013,20:846-854.
5 Teng H,Lee W Y.Optimization of Microwave-assisted Extraction of Polyphenols from Mulberry Fruits(Morusalba L.)Using Response SurfaceMethodology.JKorean Soc Appl Bio,2013,56:317-324.
6徐忠坤,陈广波,殷红梅,等.延胡索药材提取工艺研究.世界科学技术-中医药现代化,2015,17(11):2318-2321.
7陈建平,包保全,赵红梅,等.响应面优化蒙药漏芦花中总生物碱超声提取工艺研究.世界科学技术-中医药现代化,2015,17(9):1921-1928.
8袁源见,罗光明,魏春华,等.响应面优化超声波提取栀子油脂工艺研究.世界科学技术-中医药现代化,2016,18(7):1206-1211.
9刘艳杰,项荣武.星点设计效应面法在药学实验设计中的应用.中国现代应用药学杂志,2007,24(6):455-457.
10白丽,陈晓辉,徐新盛,等.高效液相色谱法测定萆薢分清丸中薯蓣皂苷元的含量.药物分析杂志,2005,25(11):1333-1335.
11 Abu-Izza K A,Garcia-Contreras L,Lu D R.Preparation and evaluation of sustained release AZT-loadedmicrospheres.2.Optimization ofmul-tiple response variables.JPharm Sci,1996,85(6):572-576.
12白丽.萆薢分清胶囊和片剂提取工艺与质量标准研究.沈阳:沈阳药科大学硕士学位论文,2005:16-18.
13姜峰,赵燕禹,姜梅兰,等.功率超声在中药提取过程中的应用.化工进展,2007,26(7):944-948.
14李博伦,周艳粉,幸一士,等.萆薢分清饮口服并保留灌肠与口服抗生素联合盐酸坦索罗辛治疗慢性前列腺炎疗效对比.吉林医学, 2013,34(14):2713-2714.
15刘嘉,赵庆年,曾明月,等.萆薢的本草考证及现代研究中医药信息,2016,33(1):120-123.
Optim ization of the Extraction Processof BiXie Fen Qing Drink Using Ultrasound-M icrowave Cooperation w ith CentralCom posite Design-Response SurfaceM ethod
Ye Yingjun,Xu Zijin,Chen Hui,Liu Jianming,Qiu Mochang
(DepartmentofPharmacy,JiangxiMedicalCollege,Shangrao 314000,China)
This study aimed at optimizing the extraction process of Bi Xie Fen Qing(BXFQ)drink using ultrasoundmicrowave cooperation with central composite design-response surface method in comparison with the traditional decoction process.Takingmicrowave frequency,amountofwater,extraction time as themain detection factors,diosgenin, glycyrrhizic acid and the extract yield were tested as the evaluation indexes;and taking an integrated score as response value for response surface and contour,predictive analysiswas carried out and the optimum extraction conditionswere achieved.Itwas found that the optimum extraction process of BXFQ drink was identified:themicrowave frequency was 434W,water addition was 18.4 times,extraction time was 9.3 mins and the ultrasonic frequency was fixed at 50W. Under the optimum process,the diosgenin extraction yield ratewas 23.17%(mg·g-1),extraction yield rate ofglycyrrhizic acid was 0.64%(g·g-1),and the extraction yield rate of extractum was 34.12%(g·g-1).All the indexeswere superior to those of the traditionalmethod.It is concluded that the composite design-response surfacemethod is suitable for the extraction optimization ofBXFQ drink with favorable predictability of themathematicalmodel.Theoptimized ultrasoundmicrowave cooperation waseasy to operatewith high extraction efficiency.It is suitable for industrialized productionwith the provision ofa scientific reference for themodern formulation developmentof BXFQ drink.
BiXie Fen Qing drink,ultrasound-microwave cooperation,centralcomposite design,extraction rate
10.11842/wst.2017.02.026
R283.3
A
(责任编辑:陈宁,责任译审:朱黎婷)
2016-11-13
修回日期:2016-12-20
*江西省教育厅科学技术研究项目(GJJ151340):中药复方温敏凝胶直肠给药系统的研究,负责人:叶颖俊。
**通讯作者:徐子金,硕士,讲师,主要研究方向:药物新剂型与新型给药系统的研究。
猜你喜欢
基层中医药(2022年2期)2022-07-22 07:39:34
天然产物研究与开发(2018年8期)2018-09-10 05:48:36
中成药(2017年9期)2017-12-19 13:34:31
中成药(2017年9期)2017-12-19 13:34:28
中国中医药信息杂志(2016年5期)2016-12-01 06:07:20
中成药(2016年4期)2016-05-17 06:08:05
测绘科学与工程(2016年4期)2016-04-17 06:51:10
医学研究杂志(2015年8期)2015-06-22 14:00:57
特产研究(2014年4期)2014-04-10 12:54:20
中成药(2014年9期)2014-02-28 22:28:51
