
基于UPLC技术测定栀子主要有效成分及特征指纹图谱研究*
2017-04-10 06:47:05李兆星申洁何春年李学刚彭勇
世界科学技术-中医药现代化 2017年2期
李兆星,申洁,何春年**,李学刚,彭勇
(1.中国医学科学院北京协和医学院药用植物研究所北京100193;2.国家教育部中草药物质基础与资源利用重点实验室北京100193;3.西南大学药学院重庆400716)
基于UPLC技术测定栀子主要有效成分及特征指纹图谱研究*
李兆星1,2,3,申洁1,2,何春年1,2**,李学刚3,彭勇1,2
(1.中国医学科学院北京协和医学院药用植物研究所北京100193;2.国家教育部中草药物质基础与资源利用重点实验室北京100193;3.西南大学药学院重庆400716)
目的:为进一步提升栀子药材的质量评价方法,该文建立了同时测定栀子药材中4个主要有效成分(京尼平苷酸、栀子苷、西红花苷-Ⅰ、西红花苷-Ⅱ)含量检测方法。方法:采用超高效液相色谱法,Waters Acquity UPLCBEH-C18色谱柱(2.1mm×100mm,1.7μm),以乙腈(A)-0.1%甲酸水溶液(B)为流动相,梯度洗脱,流速0.3mL·min-1;检测波长分别为240 nm、440 nm。在优化的色谱条件下,4个有效成分浓度在0.006 5-96μg·mL-1范围内线性关系良好,相关系数(R2)不低于0.999 5,检测限为0.038 6-0.187 5μg·mL-1。结果:对不同产地12批栀子药材的含量测定表明,不同产地栀子药材中4个成分的含量存在较大差异,其中栀子苷、西红花苷-Ⅰ含量分别为2.44%-6.96%、1.26%-3.04%。同时利用ChemPattern软件构建了12批栀子药材的特征指纹图谱,并采用多元统计方法(相似度SA、主成分分析PCA和聚类分析HCA)对不同样品测定结果进行了分析和探讨。结论:所建立的方法为栀子药材及饮片的质量评价和控制提供了参考。
栀子超高效液相色谱指纹图谱环烯醚萜类西红花苷类含量测定
栀子(Gardeniae Fructus)为茜草科植物栀子Gardenia jasminoides Ellis的干燥成熟果实,始载于《神农本草经》,并被各版中国药典收载,具有清热利湿、凉血解毒、泻火除烦、消肿止痛等功效[1]。已有大量研究表明环烯醚萜类(主要为栀子苷类化合物)和西红花苷类成分是栀子的主要有效成分[2-7],然而2015版《中国药典》(一部)栀子药材项下仅以栀子苷的含量作为含量测定控制指标,难以反映栀子药材的整体质量。当前中药质量评价方法趋向于多成分定量结合指纹图谱技术进行整体评价,目前已有一些关于栀子药材的多成分含量测定研究报道[8-13],本文在上述研究基础上进一步采用快速高效且能够多波长切换的UPLC-DAD方法,建立栀子药材中2个主要环烯醚萜类成分(京尼平苷酸、栀子苷)和2个主要西红花苷类成分(西红花苷-Ⅰ、西红花苷-Ⅱ)的含量测定方法,并借助化学和多元统计方法(SA、PCA和HCA),通过比较与分析不同产地来源的栀子样品间的差异,为完善栀子的质量评价方法提供依据和参考。
1 实验材料
1.1 仪器与试药
DIONEX U3000UPLC(包括HPG-3400RS高压泵系统,SRD-3400脱气装置,WPS-3000TRS自动进样器,TCC 3000 RS柱温箱和DAD 3000 RS检测器,数据采集与处理采用Chromeleon 7.1色谱工作站,美国Thermo Fisher),AL204型电子天平(Merrler Toledo公司),BJ-150型高速多功能粉碎机(德清拜杰电器有限公司),KQ-5200 DE型超声波清洗器(江苏宏凯仪器厂),Milli-QAcademic A10超纯水系统(美国Millipore公司)。
京尼平苷酸、栀子苷、西红花苷-Ⅰ、西红花苷-Ⅱ(成都普瑞法科技开发有限公司,生产批号:15051706、15051711、15061804、15061805),经超高效液相色谱以归一化法检验,纯度均大于98%。乙腈、甲醇、乙醇(色谱纯,美国Honeywell公司),甲酸、乙酸(色谱纯,德国CNW公司),超纯水(自制)。
1.2 实验样品
从各地药材市场(药店)或产地共收购和采集了12个省、市(区)的生栀子12种(表1),经中国医学科学院药用植物研究所彭勇研究员鉴定为茜草科植物栀子Gardenia jasminoides Ellis的干燥成熟果实,全部样品均保存于药用植物研究所亲缘中心。将各药材粉碎,过60目筛,备用。
2 方法与结果
2.1 对照品及供试品溶液的制备
2.1.1 混合对照品储备液
称取京尼平苷酸、栀子苷、西红花苷-Ⅰ、西红花苷-Ⅱ的对照品适量,精密称定,置于同一容量瓶中,以甲醇溶解,制得含上述对照品2.08、96.00、104.58、8.16μg·mL-1的混合溶液,即得。
2.1.2 供试品溶液
精密称取栀子药材粉末(过60目筛)0.05 g,置于50mL容量瓶,精密加入甲醇50mL,称重,超声处理(功率200W,频率40 kHz,温度40℃)30min。放冷后,称重,以甲醇补足失重后,摇匀,用0.22μm微孔滤膜滤过,取续滤液,即得。
2.2 色谱条件
色谱柱:Waters Acquity UPLC BEH-C18色谱柱(2.1mm×100mm,1.7μm);流动相:乙腈(A)-0.1%甲酸水溶液(B),梯度洗脱(0-2min 2%A,4-5min 8% A,6min 13%A,8min 17.5%A,9min 24%A,11-12min 25%A,13min 30%A,14min 33%A,15-17min 35% A,20min 70%A),检测波长为240 nm,440 nm,流速0.3mL·min-1,柱温度40℃,进样量1μL。在上述色谱条件中,主要组分与相邻峰分离度均大1.5,出峰时间也得到了很好的控制,见图1,其中图中0-10min是240 nm波长检测下的色谱图,10-20min是440 nm波长检测下的色谱图。
表1 不同产地的栀子药材样品
2.3 线性关系考察
精密吸取混合对照品溶液,不断稀释至浓度为原溶液浓度的1/2,依次进样至超高效液相色谱仪,进样量1μL。分别以京尼平苷酸、栀子苷、西红花苷-Ⅰ、西红花苷-Ⅱ的进样浓度(μg·mL-1)为横坐标,峰面积积分值(mAU·min-1)为纵坐标绘制标准曲线,计算回归方程,结果见表2,结果表明4个化合物均有较好的线性关系。同时测定最低检测限(LOD),即仪器的信噪比S/N等于3,最低定量限(LOQ),即仪器的信噪比S/N等于10,结果见表2。
2.4 精密度试验
精密吸取批号为SZZ01样品的供试品溶液,连续进样6次,记录峰面积,计算得4个成分峰面积的RSD依次为0.77%、1.34%、0.63%、2.34%,均小于3.0%,表明仪器精密度良好。
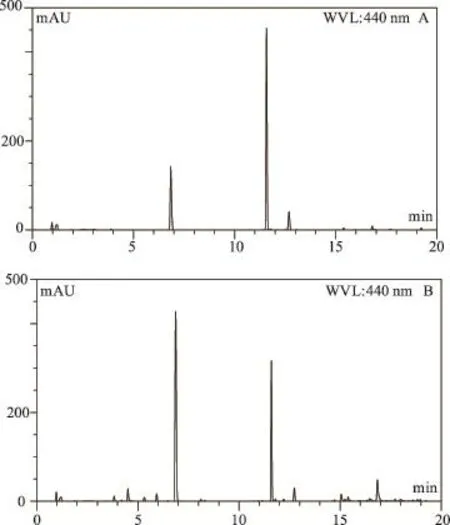
图1 混合对照品(A)和栀子样品(B)的UPLC图
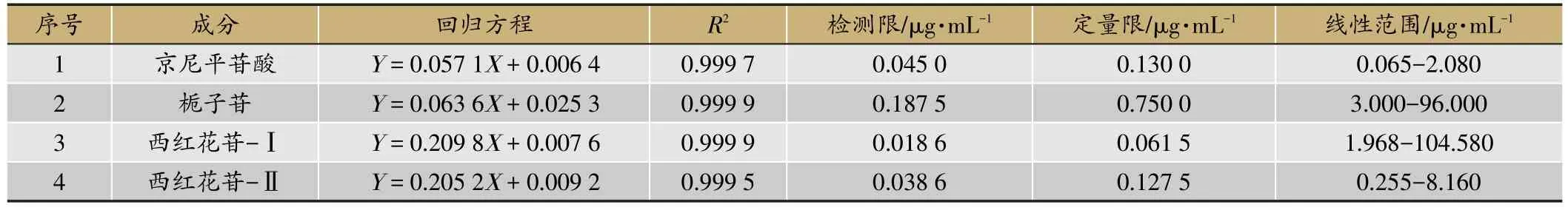
表2 线性关系考察
2.5 稳定性试验
精密称取批号为SZZ01样品0.05 g,以“2.1.2”项下方法制得供试品溶液。在上述色谱条件下分别在0、1、2、3、4、5、6、12、24 h依法进样测定,记录峰面积,计算得4个成分峰面积的RSD依次为2.24%、2.59%、2.28%、1.36%,均小于3.0%,表明供试品溶液在24 h内稳定。
2.6 重复性试验
精密称取批号为SZZ01的样品0.05 g,共6份,分别以“2.1.2”项下方法平行制备供试品溶液,进样1μL,记录峰面积,计算得4个成分峰面积的RSD依次为2.31%、1.28%、1.63%、2.77%,均小于3.0%,表明该方法重复性良好。
2.7 回收率试验
精密称取批号为SZZ01的样品9份,每份0.05 g,并分别以原供试品溶液中含量的80%、100%、120%精密加入上述4个对照品适量。分别以“2.1.2”项下方法平行制备供试品溶液,在上述色谱条件下,进样1μL,记录色谱峰面积,并计算回收率。结果见表3。京尼平苷酸、栀子苷、西红花苷-Ⅰ、西红花苷-Ⅱ的平均回收率分别为100.5%、99.9%、100.9%、99.5%。说明该方法准确度较高。
2.8 样品测定
精密称取表1中12份栀子药材粉末各0.05 g,并分别以“2.1.2”项下方法平行制备供试品溶液,进样1μL,记录峰面积,以“2.3”项下回归方程计算各有效成分的含量。结果见表4,图2。
2.9 特征指纹图谱的建立
2.9.1 指纹图谱共有模式的建立
将12批样品分析结果导入ChemPattern软件(旗舰版,科迈恩科技有限公司),选择浙江产栀子药材(SZZ01)为参照图谱,进行多图谱手动匹配来消除色谱峰保留时间的漂移,总共得出13个峰,进行过滤去除一些小杂峰后得到栀子药材的特征指纹图谱,图中共有9个明显的色谱峰,根据保留时间和紫外吸收图谱特征,鉴别出峰号2、5、6和7分别为京尼平苷酸、栀子苷、西红花苷-Ⅰ、西红花苷-Ⅱ。结果见图3。12批栀子药材色谱对比图谱见图4。
2.9.2 相似度评价
采用ChemPattern软件对12批栀子药材的UPLC指纹图谱进行相似度评价,采用余弦夹角法计算,结果见表5。分析结果显示有11批次的栀子相似度均在0.97以上,具有较高的相似度。SZZ03相似度相对较低,但仍高于0.900,表明各产地栀子没有显著差异。其主要差异表现在有效活性成分环烯醚萜类(栀子苷)和西红花苷类(西红花苷-I)的含量上。
2.9.3 主成分分析
运用主成分分析法[14],得出,三个主成分PC1、PC2和PC3,总共能解释98.63%的总变异,分析结果如图5、图6。

表3 加样回收率试验(n=3)
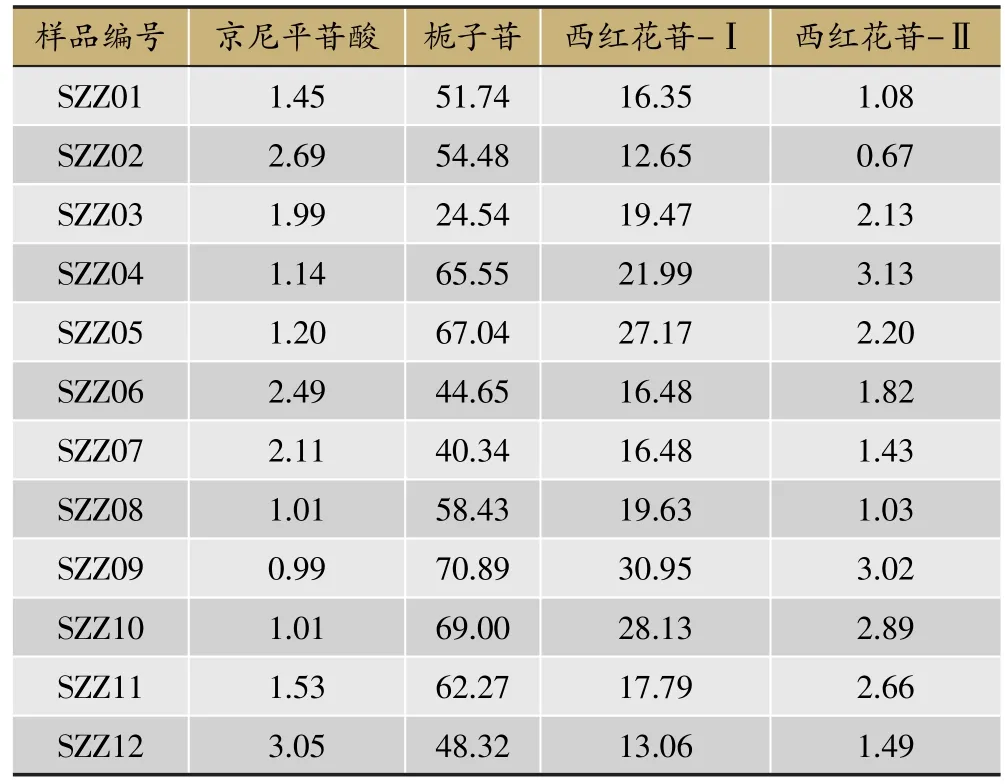
表4 栀子药材中4种有效成分的含量/mg·g-1
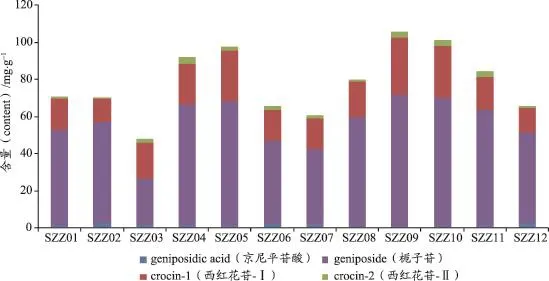
图2 栀子药材中4种有效成分的含量图
由图5可以看出12批栀子药材主成分得分分布相互重叠,其化学特征比较相似,没有显示出明显差异;各组内样品分布较为分散,可能与样品之间化学成分含量差异较大有关。由图6可以看出,变量5(栀子苷),变量6(西红花苷-Ⅰ)和变量7(西红花苷-Ⅱ)对主成分得分结果有较大的影响。其结果表明造成不同样品间差异的主要因素可能是环烯醚萜类成分栀子苷和西红花苷类成分西红花苷-Ⅰ及西红花苷-Ⅱ含量以及比例的差异。
2.9.4 聚类分析
根据化学成分对栀子进行UPGMA聚类分析,得到12批栀子的聚类分析图(图7)。12批栀子可分为4个分支:①产地为浙江、四川宜宾、江西赣州、江西抚州、湖南邵阳的五批栀子聚为一类;②产地为四川绵阳、山西、安徽毫州、广西玉林、广东茂名的五批栀子聚为一类;③产地为河南南阳、江西宜春的栀子各自为一类。总之不同地区的栀子可聚到一类,同一地区的栀子又聚类到不同类,其原因可能与种植条件、采收时间有关。

图3 栀子药材共有模式图

图4 12批栀子药材UPLC指纹图谱

表5 12批栀子药材的相似度分析
3 讨论
3.1 栀子样品提取条件的优化与确定
栀子药材主要含有环烯醚萜类和西红花苷类两类有效成分,其中栀子苷和西红花苷-Ⅰ这两种成分在药材中含量高,紫外吸收系数大,为了确保高含量成分充分溶出,并适宜检测,本实验确定药材粉末取样量为0.05 g,提取溶剂体积为50mL。由于环烯醚萜类和西红花苷类成分为中等极性和较大极性成分,根据文献报道[15,16]两类成分均易溶于甲醇、乙醇等极性溶剂,同时考虑到甲醇、乙醇的低沸点。因此本文选择水、50%甲醇、甲醇及50%乙醇、乙醇为溶剂,分别采取超声、60℃温水浴加热和水浴回流等提取方法,并考察不同时间的提取效率。结果表明以甲醇为提取溶剂提取效果最好,超声提取与水浴加热提取率无明显差异,提取时间30min与60min提取率无明显差异,因此本实验确定提取方法为:精密称取栀子药材粉末0.05 g,加入甲醇50mL,超声提取30min。
3.2 流动相选择
分别采用甲醇-水,乙腈-水,乙腈-0.1%甲酸水溶液,乙腈-0.1%乙酸水溶液,乙腈-0.2%乙酸水溶液,乙腈-0.3%乙酸水溶液,乙腈-0.2%甲酸水溶液,乙腈-0.3%甲酸水溶液进行不同等度洗脱和线性梯度洗脱。结果表明,在乙腈-0.1%甲酸水溶液的流动相中,各色谱峰的分布、峰形和分离度达到最佳状态,因此,选择乙腈-0.1%甲酸水溶液作为栀子药材超高效液相色谱分析的流动相。
3.3 测定波长选择
环烯醚萜类、西红花苷类的最大吸收波长分别为240 nm和440 nm,因此本实验在含量测定中确定240 nm为京尼平苷酸和栀子苷的检测波长,440 nm为西红花苷-Ⅰ和西红花苷-Ⅱ的检测波长[17]。在特征指纹图谱提取中,考虑到环烯醚萜类成分主要在前10min出峰,而西红花苷类成分集中在10min后,故本实验在10min时设置了波长变换,0-10min采用240 nm检测,10-20min采用440 nm波长检测。
3.4 含量测定结果分析
2015年版《中国药典》一部栀子药材及其饮片项下,均以栀子苷(即京尼平苷)作为指标性成分,并规定含量不得少于1.8%[1],本文对12个不同来源的栀子药材含量测定结果表明:栀子苷的含量最高,12份药材的含量范围为24.54-70.89mg·g-1,均明显高于药典规定,其中样品SZZ04、SZZ05、SZZ09、SZZ10和SZZ11中栀子苷含量较高,均大于62.27mg·g-1(即6.227%),仅样品SZZ03(产地为江西宜春)的含量稍低,也达到2.454%;京尼平苷酸为栀子苷分子中酯键水解脱去甲基的产物,在12份药材的含量范围为0.99-3.05mg·g-1,在栀子药材中含量明显低于栀子苷一个数量级以上,相对来说样品SZZ12中京尼平苷酸含量较高,达到0.304%;西红花苷-Ⅰ为栀子药材中含量最高水溶性色素,在12份样品中含量为12.65-30.95mg·g-1,其中样品SZZ05、SZZ09和SZZ10中含量相对较高,均大于27.17mg·g-1(即2.717%);西红花苷-Ⅱ为西红花苷-Ⅰ分子中脱去一个糖,在栀子中含量比西红花苷-Ⅰ低约一个数量级,含量范围为0.67-3.13mg·g-1,其中在样品SZZ04、SZZ09和SZZ10中含量相对较高,均大于2.88mg·g-1(即0.288%)。
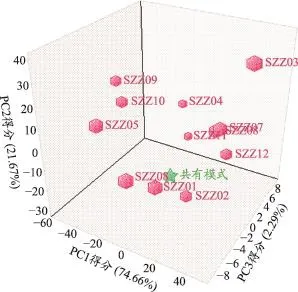
图5 12批栀子药材的主成分分析得分图
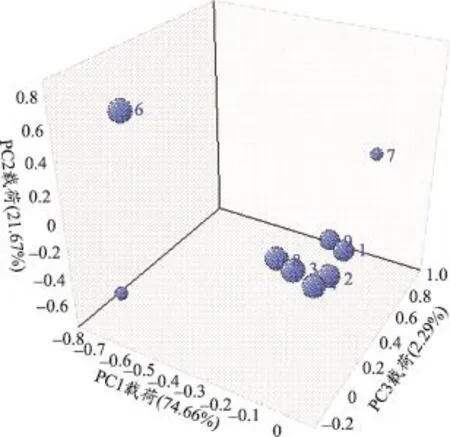
图6 12批栀子药材的主成分分析载荷图
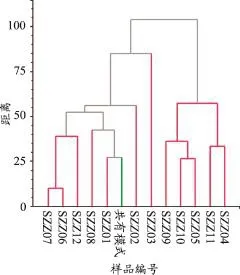
图7 12批栀子药材的UPGMA聚类图
4 小结
本文建立了同时测定栀子药材中4个主要有效成分(京尼平苷酸、栀子苷、西红花苷-Ⅰ和西红花苷-Ⅱ)含量的UPLC-DAD方法,对不同产地12批栀子药材的测定结果表明:4种有效成分含量差异较大,如分别产自山西、安徽、广西产的3份样品中栀子苷和西红花苷-Ⅰ含量均较高,提示目前市场流通的栀子药材质量的均一性不够理想,对药材的临床疗效可能会产生较大影响,需要进一步深入研究。同时构建了12批栀子药材的特征指纹图谱,并采用多元统计方法(相似度、主成分分析和聚类分析)对不同样品测定结果进行了分析和探讨。所建立的方法为栀子药材及饮片的质量评价和控制提供了参考。
1国家药典委员会.中华人民共和国药典(2015年版一部).北京:中国医药科技出版社,2015:248.
2于洋,高昊,戴毅,等.栀子属植物化学成分的研究进展.中草药, 2010,44(1):148-153.
3雷婷婷,张海燕,刘星星,等.栀子中4种环烯醚萜苷类成分治疗大鼠血管性痴呆的药效学研究.中国新药杂志,2013,22(21):2487-2491.
4张海燕,邬伟魁,杨军宣,等.栀子对心脑血管系统的作用研究.中国实验方剂学杂志,2011,17(14):294-298.
5 Park JH,Yoon J,Lee K Y,etal.Effectsofgeniposideon hepatocytesundergoing epithelial-mesenchymal transition in hepatic fibrosisby targeting TGFβ/Smad and ERK-MAPK signaling pathways.Biochimie,2015, 113:26-34.
6 Hoshyar R,Bathaie SZ,Sadeghizadeh M.Crocin triggers the apoptosis through increasing the Bax/Bcl-2 ratio and caspase activation in human gastric adenocarcinoma,AGS,cells.DNACell Biol,2013,32(2):50-57.
7 Xia D.Ovarian cancer HO-8910 cell apoptosis induced by crocin in vitro.NatProd Commun,2015,10(2):249-252.
8付小梅,彭水梅,刘婧,等.HPLC法同时测定栀子类药材中10个主要有效成分的含量.药物分析杂志,2014,34(4):615-621.
9王莎,周小琴,司徒少金,等.栀子药材环烯醚萜类和西红花酸类成分HPLC指纹图谱研究.中国实验方剂学杂志,2012,18(19):85-88.
10刘武占,范建伟,高艳红,等.HPLC同时测定栀子中8个环烯醚萜苷类成分的含量.中国中药杂志,2012,37(16):2417.
11韩建萍,陈士林,张文生,等.不同产地栀子药材HPLC指纹图谱研究.世界科学技术-中医药现代化,2007,9(4):56-60.
12李云,吴建雄,石伟,等.HPLC同时测定栀子药材中两类活性成分.世界科学技术-中医药现代化,2015,17(11):2229-2234.
13米慧娟,王永香,张庆芬,等.江西省樟树县GAP基地不同采收时间栀子药材的质量研究.世界科学技术-中医药现代化,2016,18(8):1393-1400.
14胡震,杨广德,罗国安,等.栀子中栀子苷提取工艺及HPLC分析.中成药,2006,28(3):336-338.
15 Liu S,Liang Y Z,Liu H T.Chemometrics applied to quality control and metabolomics for traditional Chinesemedicines.JChromatogr B Analyt TechnolBiomed Life Sci,2016,1015-1016:82-91.
16黄弦,左月明,罗光明,等.多指标评价栀子超声提取工.时珍国医国药,2013,24(7):1568-1571.
17蔡少青,李军.常用中药材品种整理和质量研究(北方编第5册).北京:北京医科大学出版社,2001:278-317.
The Determ ination ofM ajor Effective Com pounds in Gardeniae Fructus from Different Regions and the Establishmentof Fingerprints Based on UPLC
LiZhaoxing1,2,3,Shen Jie1,2,HeChunnian1,2,LiXuegang3,Peng Yong1,2
(1.Institute ofMedicinal PlantDevelopment,Chinese Academy ofMedical Science,Peking Union MedicalCollege, Beijing 100193,China;2.Key Laboratory ofBioactive Substancesand ResourcesUtilization of ChineseHerbalMedicine,Ministry ofEducation,Beijing 10019,China;3.SchoolofPharmaceutical Sciences, SouthwestUniversity,Chongqing 400715,China)
For promoting the quality evaluation of Gardenia resource,a UPLC method for the determination of 4 compounds(geniposidic acid,geniposide,crocin-1 and crocin-2)in Gardeniae Fructus was established.The UPLC separation was performed on an Waters Acquity UPLC BEH-C18(2.1 mm×100 mm,1.7μm)column eluted with the mobile phasesofacetonitrile-0.1%formic acid solution in a gradientmode ata flow rate of0.3mL·min-1.The detection wavelengths were 240 nm and 440 nm,respectively.Under the optimum conditions,the calibration curves of four analysiswere linear in the range of0.0065-0.0096μg·mL-1,with correlation coefficientsmore than 0.9995.The limitsof detection(LODs)of four analysis were in the range of 0.0386-0.1875μg·mL-1.As the results of determination of 4 compounds of 12 batches of Gardeniae Fructus showed,there was great differences between the contents of the 4 compounds,the contents of geniposide were 2.44%-6.96%,while the contents of crocin-1 were 1.26%-3.04%.In addition,fingerprints of 12 batches of Gardenia resources were built using ChemPattern software,and analysis and exploration over the detection results of several sampleswere carried outusingmultivariate statisticalmethod(similarity analysis,principal component analysis and cluster analysis).The present study provided a reference to value the importanceof thismethod in the quality evaluation and quality controlofGardeniae resourcesand slices.
Gardenia Fructus,ultra-performance liquid chromatography,fingerprint,iridoids,crocins,quantitativeanalysis
10.11842/wst.2017.02.025
R283
A
(责任编辑:王慧慧,责任译审:朱黎婷)
2016-11-22
修回日期:2017-01-13
*中国医学科学院医学与健康科技创新工程经费资助项目(2016-I2M-1-012)。
**通讯作者:何春年,副研究员,硕士/博士生导师,主要研究方向:中药及天然药物药效物质基础研究及质量评价。
猜你喜欢
煤化工(2022年3期)2022-07-08 07:24:42
快乐语文(2021年34期)2022-01-18 06:04:10
快乐语文(2021年27期)2021-11-24 01:29:16
快乐语文(2021年11期)2021-07-20 07:41:40
快乐语文(2021年15期)2021-06-15 10:19:34
基层中医药(2018年7期)2018-12-06 09:25:50
中国现代中药(2018年11期)2018-11-19 05:24:08
金色年华(2016年11期)2016-02-28 01:42:20
中国资源综合利用(2016年10期)2016-01-22 08:36:09
传奇故事(破茧成蝶)(2015年1期)2015-02-28 09:26:18
